{"title":"Wfold: A new method for predicting RNA secondary structure with deep learning","authors":"Yongna Yuan, Enjie Yang, Ruisheng Zhang","doi":"10.1016/j.compbiomed.2024.109207","DOIUrl":null,"url":null,"abstract":"<div><div>Precise estimations of RNA secondary structures have the potential to reveal the various roles that non-coding RNAs play in regulating cellular activity. However, the mainstay of traditional RNA secondary structure prediction methods relies on thermos-dynamic models via free energy minimization, a laborious process that requires a lot of prior knowledge. Here, RNA secondary structure prediction using Wfold, an end-to-end deep learning-based approach, is suggested. Wfold is trained directly on annotated data and base-pairing criteria. It makes use of an image-like representation of RNA sequences, which an enhanced U-net incorporated with a transformer encoder can process effectively. Wfold eventually increases the accuracy of RNA secondary structure prediction by combining the benefits of self-attention mechanism's mining of long-range information with U-net's ability to gather local information. We compare Wfold's performance using RNA datasets that are within and across families. When trained and evaluated on different RNA families, it achieves a similar performance as the traditional methods, but dramatically outperforms the state-of-the-art methods on within-family datasets. Moreover, Wfold can also reliably forecast pseudoknots. The findings imply that Wfold may be useful for improving sequence alignment, functional annotations, and RNA structure modeling.</div></div>","PeriodicalId":10578,"journal":{"name":"Computers in biology and medicine","volume":"182 ","pages":"Article 109207"},"PeriodicalIF":6.3000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computers in biology and medicine","FirstCategoryId":"5","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0010482524012927","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/27 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
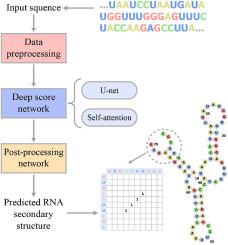
Precise estimations of RNA secondary structures have the potential to reveal the various roles that non-coding RNAs play in regulating cellular activity. However, the mainstay of traditional RNA secondary structure prediction methods relies on thermos-dynamic models via free energy minimization, a laborious process that requires a lot of prior knowledge. Here, RNA secondary structure prediction using Wfold, an end-to-end deep learning-based approach, is suggested. Wfold is trained directly on annotated data and base-pairing criteria. It makes use of an image-like representation of RNA sequences, which an enhanced U-net incorporated with a transformer encoder can process effectively. Wfold eventually increases the accuracy of RNA secondary structure prediction by combining the benefits of self-attention mechanism's mining of long-range information with U-net's ability to gather local information. We compare Wfold's performance using RNA datasets that are within and across families. When trained and evaluated on different RNA families, it achieves a similar performance as the traditional methods, but dramatically outperforms the state-of-the-art methods on within-family datasets. Moreover, Wfold can also reliably forecast pseudoknots. The findings imply that Wfold may be useful for improving sequence alignment, functional annotations, and RNA structure modeling.
期刊介绍:
Computers in Biology and Medicine is an international forum for sharing groundbreaking advancements in the use of computers in bioscience and medicine. This journal serves as a medium for communicating essential research, instruction, ideas, and information regarding the rapidly evolving field of computer applications in these domains. By encouraging the exchange of knowledge, we aim to facilitate progress and innovation in the utilization of computers in biology and medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
