Philip J Freda, Suyu Ye, Robert Zhang, Jason H Moore, Ryan J Urbanowicz
{"title":"Assessing the limitations of relief-based algorithms in detecting higher-order interactions.","authors":"Philip J Freda, Suyu Ye, Robert Zhang, Jason H Moore, Ryan J Urbanowicz","doi":"10.1186/s13040-024-00390-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Epistasis, the interaction between genetic loci where the effect of one locus is influenced by one or more other loci, plays a crucial role in the genetic architecture of complex traits. However, as the number of loci considered increases, the investigation of epistasis becomes exponentially more complex, making the selection of key features vital for effective downstream analyses. Relief-Based Algorithms (RBAs) are often employed for this purpose due to their reputation as \"interaction-sensitive\" algorithms and uniquely non-exhaustive approach. However, the limitations of RBAs in detecting interactions, particularly those involving multiple loci, have not been thoroughly defined. This study seeks to address this gap by evaluating the efficiency of RBAs in detecting higher-order epistatic interactions. Motivated by previous findings that suggest some RBAs may rank predictive features involved in higher-order epistasis negatively, we explore the potential of absolute value ranking of RBA feature weights as an alternative approach for capturing complex interactions. In this study, we assess the performance of ReliefF, MultiSURF, and MultiSURFstar on simulated genetic datasets that model various patterns of genotype-phenotype associations, including 2-way to 5-way genetic interactions, and compare their performance to two control methods: a random shuffle and mutual information.</p><p><strong>Results: </strong>Our findings indicate that while RBAs effectively identify lower-order (2 to 3-way) interactions, their capability to detect higher-order interactions is significantly limited, primarily by large feature count but also by signal noise. Specifically, we observe that RBAs are successful in detecting fully penetrant 4-way XOR interactions using an absolute value ranking approach, but this is restricted to datasets with only 20 total features.</p><p><strong>Conclusions: </strong>These results highlight the inherent limitations of current RBAs and underscore the need for the development of Relief-based approaches with enhanced detection capabilities for the investigation of epistasis, particularly in datasets with large feature counts and complex higher-order interactions.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"17 1","pages":"37"},"PeriodicalIF":6.1000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11443793/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-024-00390-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Epistasis, the interaction between genetic loci where the effect of one locus is influenced by one or more other loci, plays a crucial role in the genetic architecture of complex traits. However, as the number of loci considered increases, the investigation of epistasis becomes exponentially more complex, making the selection of key features vital for effective downstream analyses. Relief-Based Algorithms (RBAs) are often employed for this purpose due to their reputation as "interaction-sensitive" algorithms and uniquely non-exhaustive approach. However, the limitations of RBAs in detecting interactions, particularly those involving multiple loci, have not been thoroughly defined. This study seeks to address this gap by evaluating the efficiency of RBAs in detecting higher-order epistatic interactions. Motivated by previous findings that suggest some RBAs may rank predictive features involved in higher-order epistasis negatively, we explore the potential of absolute value ranking of RBA feature weights as an alternative approach for capturing complex interactions. In this study, we assess the performance of ReliefF, MultiSURF, and MultiSURFstar on simulated genetic datasets that model various patterns of genotype-phenotype associations, including 2-way to 5-way genetic interactions, and compare their performance to two control methods: a random shuffle and mutual information.
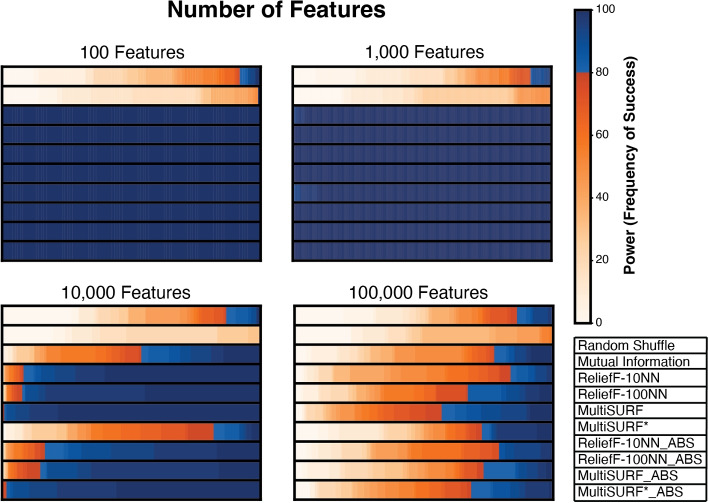
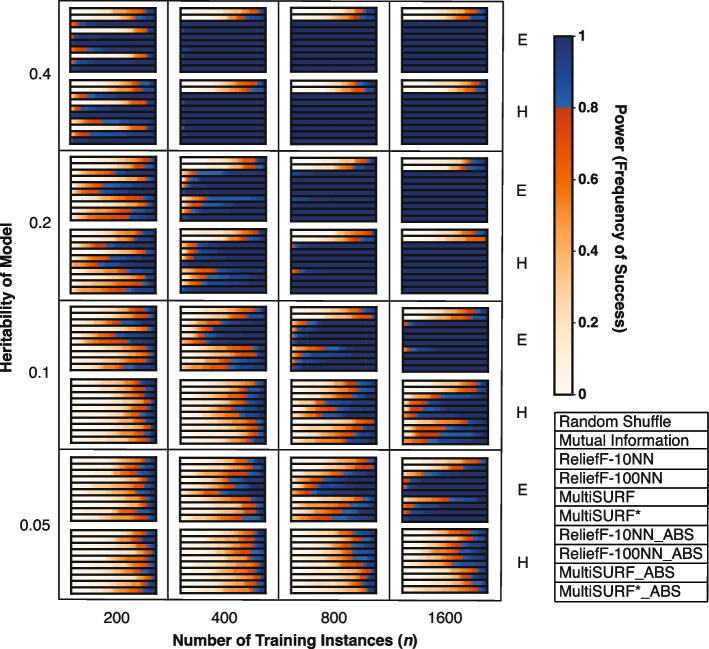
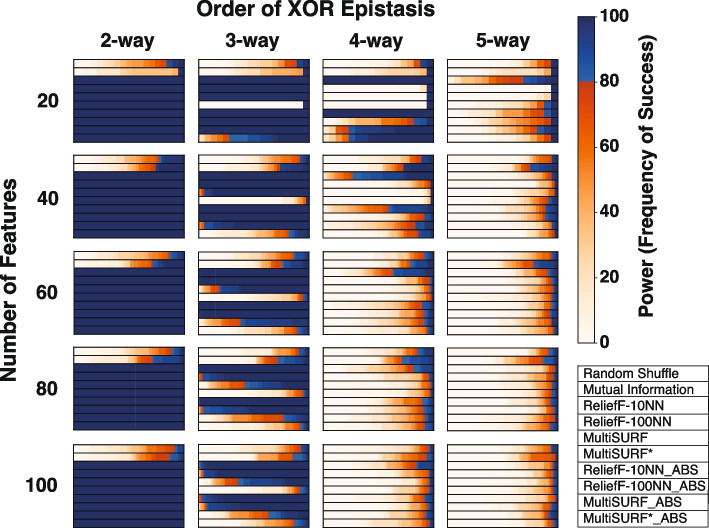
Results: Our findings indicate that while RBAs effectively identify lower-order (2 to 3-way) interactions, their capability to detect higher-order interactions is significantly limited, primarily by large feature count but also by signal noise. Specifically, we observe that RBAs are successful in detecting fully penetrant 4-way XOR interactions using an absolute value ranking approach, but this is restricted to datasets with only 20 total features.
Conclusions: These results highlight the inherent limitations of current RBAs and underscore the need for the development of Relief-based approaches with enhanced detection capabilities for the investigation of epistasis, particularly in datasets with large feature counts and complex higher-order interactions.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
