{"title":"Diagnostic options, physiopathology, risk factors and genetic causes of permanent congenital hypothyroidism: A narrative review.","authors":"Zahra Rasoulizadeh, Mahtab Ordooei, Elahe Akbarian","doi":"10.22088/cjim.15.4.570","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>In Permanent congenital hypothyroidism (PCH) is a lifelong condition characterized by a deficiency in thyroid hormone, leading to various neurodevelopmental complications. Early clinical signs are often nonspecific and easily overlooked, but newborn screening programs have improved early detection.</p><p><strong>Methods: </strong>This narrative review aims to provide insights comparatively transient and permanent PCH and also the diagnosis, risk factors, underlying pathophysiology, and genetic causes associated with PCH. Relevant studies were identified through a comprehensive search using the term 'Permanent congenital hypothyroidism' (Mesh) across scientific databases of electronic databases such as PubMed, Scopus, and Web of Science.</p><p><strong>Results: </strong>Prompt initiation of thyroid hormone replacement therapy, particularly within the initial two weeks postpartum, crucially enhances neurocognitive development outcomes. Multiple predictive approaches, encompassing screening TSH levels, maternal thyroid history, and levothyroxine dosage per kilogram assessment, aid in identifying PCH. Recent studies have demonstrated a mounting prevalence of PCH, contributing significantly to the overall rise in CH incidence. Genetic factors, primarily DUOX2 and DUOXA2 mutations, alongside environmental influences such as post-term birth, low birth weight, and macrosomia, may induce PCH. Nonetheless, reliable markers for early PCH prediction upon diagnosis remain elusive, leading to delayed recognition post-ceasing levothyroxine treatment around age 3.</p><p><strong>Conclusions: </strong>Recent studies have observed an increased incidence of PCH, contributing substantially to the overall rise in cases of congenital hypothyroidism. Understanding the diagnostic options and genetic etiologies associated with PCH is crucial for the early identification and appropriate management.</p>","PeriodicalId":9646,"journal":{"name":"Caspian Journal of Internal Medicine","volume":"15 4","pages":"570-578"},"PeriodicalIF":1.0000,"publicationDate":"2024-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11444113/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Caspian Journal of Internal Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.22088/cjim.15.4.570","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
Background: In Permanent congenital hypothyroidism (PCH) is a lifelong condition characterized by a deficiency in thyroid hormone, leading to various neurodevelopmental complications. Early clinical signs are often nonspecific and easily overlooked, but newborn screening programs have improved early detection.
Methods: This narrative review aims to provide insights comparatively transient and permanent PCH and also the diagnosis, risk factors, underlying pathophysiology, and genetic causes associated with PCH. Relevant studies were identified through a comprehensive search using the term 'Permanent congenital hypothyroidism' (Mesh) across scientific databases of electronic databases such as PubMed, Scopus, and Web of Science.
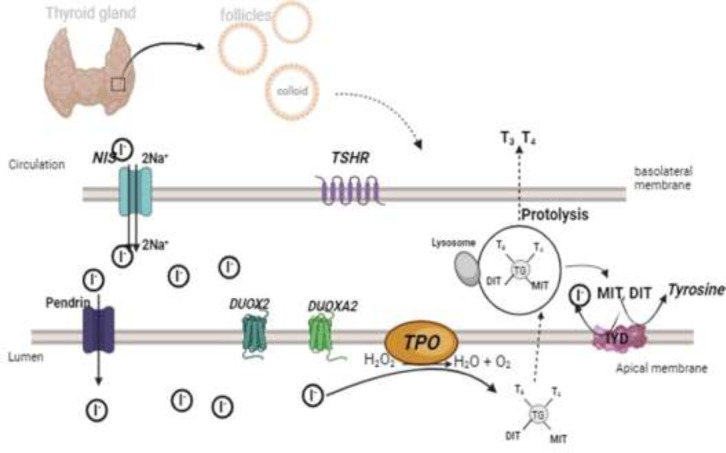
Results: Prompt initiation of thyroid hormone replacement therapy, particularly within the initial two weeks postpartum, crucially enhances neurocognitive development outcomes. Multiple predictive approaches, encompassing screening TSH levels, maternal thyroid history, and levothyroxine dosage per kilogram assessment, aid in identifying PCH. Recent studies have demonstrated a mounting prevalence of PCH, contributing significantly to the overall rise in CH incidence. Genetic factors, primarily DUOX2 and DUOXA2 mutations, alongside environmental influences such as post-term birth, low birth weight, and macrosomia, may induce PCH. Nonetheless, reliable markers for early PCH prediction upon diagnosis remain elusive, leading to delayed recognition post-ceasing levothyroxine treatment around age 3.
Conclusions: Recent studies have observed an increased incidence of PCH, contributing substantially to the overall rise in cases of congenital hypothyroidism. Understanding the diagnostic options and genetic etiologies associated with PCH is crucial for the early identification and appropriate management.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
