Jun Hyeong Park, June Hyuck Lim, Seonhwa Kim, Chul-Ho Kim, Jeong-Seok Choi, Jun Hyeok Lim, Lucia Kim, Jae Won Chang, Dongil Park, Myung-won Lee, Sup Kim, Il-Seok Park, Seung Hoon Han, Eun Shin, Jin Roh, Jaesung Heo
{"title":"Deep learning-based analysis of EGFR mutation prevalence in lung adenocarcinoma H&E whole slide images","authors":"Jun Hyeong Park, June Hyuck Lim, Seonhwa Kim, Chul-Ho Kim, Jeong-Seok Choi, Jun Hyeok Lim, Lucia Kim, Jae Won Chang, Dongil Park, Myung-won Lee, Sup Kim, Il-Seok Park, Seung Hoon Han, Eun Shin, Jin Roh, Jaesung Heo","doi":"10.1002/2056-4538.70004","DOIUrl":null,"url":null,"abstract":"<p><i>EGFR</i> mutations are a major prognostic factor in lung adenocarcinoma. However, current detection methods require sufficient samples and are costly. Deep learning is promising for mutation prediction in histopathological image analysis but has limitations in that it does not sufficiently reflect tumor heterogeneity and lacks interpretability. In this study, we developed a deep learning model to predict the presence of <i>EGFR</i> mutations by analyzing histopathological patterns in whole slide images (WSIs). We also introduced the <i>EGFR</i> mutation prevalence (EMP) score, which quantifies <i>EGFR</i> prevalence in WSIs based on patch-level predictions, and evaluated its interpretability and utility. Our model estimates the probability of EGFR prevalence in each patch by partitioning the WSI based on multiple-instance learning and predicts the presence of <i>EGFR</i> mutations at the slide level. We utilized a patch-masking scheduler training strategy to enable the model to learn various histopathological patterns of EGFR. This study included 868 WSI samples from lung adenocarcinoma patients collected from three medical institutions: Hallym University Medical Center, Inha University Hospital, and Chungnam National University Hospital. For the test dataset, 197 WSIs were collected from Ajou University Medical Center to evaluate the presence of <i>EGFR</i> mutations. Our model demonstrated prediction performance with an area under the receiver operating characteristic curve of 0.7680 (0.7607–0.7720) and an area under the precision-recall curve of 0.8391 (0.8326–0.8430). The EMP score showed Spearman correlation coefficients of 0.4705 (<i>p</i> = 0.0087) for p.L858R and 0.5918 (<i>p</i> = 0.0037) for exon 19 deletions in 64 samples subjected to next-generation sequencing analysis. Additionally, high EMP scores were associated with papillary and acinar patterns (<i>p</i> = 0.0038 and <i>p</i> = 0.0255, respectively), whereas low EMP scores were associated with solid patterns (<i>p</i> = 0.0001). These results validate the reliability of our model and suggest that it can provide crucial information for rapid screening and treatment plans.</p>","PeriodicalId":48612,"journal":{"name":"Journal of Pathology Clinical Research","volume":"10 6","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2024-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11446692/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Pathology Clinical Research","FirstCategoryId":"3","ListUrlMain":"https://pathsocjournals.onlinelibrary.wiley.com/doi/10.1002/2056-4538.70004","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PATHOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
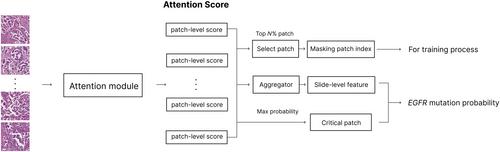
EGFR mutations are a major prognostic factor in lung adenocarcinoma. However, current detection methods require sufficient samples and are costly. Deep learning is promising for mutation prediction in histopathological image analysis but has limitations in that it does not sufficiently reflect tumor heterogeneity and lacks interpretability. In this study, we developed a deep learning model to predict the presence of EGFR mutations by analyzing histopathological patterns in whole slide images (WSIs). We also introduced the EGFR mutation prevalence (EMP) score, which quantifies EGFR prevalence in WSIs based on patch-level predictions, and evaluated its interpretability and utility. Our model estimates the probability of EGFR prevalence in each patch by partitioning the WSI based on multiple-instance learning and predicts the presence of EGFR mutations at the slide level. We utilized a patch-masking scheduler training strategy to enable the model to learn various histopathological patterns of EGFR. This study included 868 WSI samples from lung adenocarcinoma patients collected from three medical institutions: Hallym University Medical Center, Inha University Hospital, and Chungnam National University Hospital. For the test dataset, 197 WSIs were collected from Ajou University Medical Center to evaluate the presence of EGFR mutations. Our model demonstrated prediction performance with an area under the receiver operating characteristic curve of 0.7680 (0.7607–0.7720) and an area under the precision-recall curve of 0.8391 (0.8326–0.8430). The EMP score showed Spearman correlation coefficients of 0.4705 (p = 0.0087) for p.L858R and 0.5918 (p = 0.0037) for exon 19 deletions in 64 samples subjected to next-generation sequencing analysis. Additionally, high EMP scores were associated with papillary and acinar patterns (p = 0.0038 and p = 0.0255, respectively), whereas low EMP scores were associated with solid patterns (p = 0.0001). These results validate the reliability of our model and suggest that it can provide crucial information for rapid screening and treatment plans.
期刊介绍:
The Journal of Pathology: Clinical Research and The Journal of Pathology serve as translational bridges between basic biomedical science and clinical medicine with particular emphasis on, but not restricted to, tissue based studies.
The focus of The Journal of Pathology: Clinical Research is the publication of studies that illuminate the clinical relevance of research in the broad area of the study of disease. Appropriately powered and validated studies with novel diagnostic, prognostic and predictive significance, and biomarker discover and validation, will be welcomed. Studies with a predominantly mechanistic basis will be more appropriate for the companion Journal of Pathology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
