{"title":"Review of the technology used for structural characterization of the GMO genome using NGS data.","authors":"Kahee Moon, Prakash Basnet, Taeyoung Um, Ik-Young Choi","doi":"10.1186/s44342-024-00016-1","DOIUrl":null,"url":null,"abstract":"<p><p>The molecular characterization of genetically modified organisms (GMOs) is essential for ensuring safety and gaining regulatory approval for commercialization. According to CODEX standards, this characterization involves evaluating the presence of introduced genes, insertion sites, copy number, and nucleotide sequence structure. Advances in technology have led to the increased use of next-generation sequencing (NGS) over traditional methods such as Southern blotting. While both methods provide high reproducibility and accuracy, Southern blotting is labor-intensive and time-consuming due to the need for repetitive probe design and analyses for each target, resulting in low throughput. Conversely, NGS facilitates rapid and comprehensive analysis by mapping whole-genome sequencing (WGS) data to plasmid sequences, accurately identifying T-DNA insertion sites and flanking regions. This advantage allows for efficient detection of T-DNA presence, copy number, and unintended gene insertions without additional probe work. This paper reviews the current status of GMO genome characterization using NGS and proposes more efficient strategies for this purpose.</p>","PeriodicalId":94288,"journal":{"name":"Genomics & informatics","volume":"22 1","pages":"14"},"PeriodicalIF":0.0000,"publicationDate":"2024-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11445869/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics & informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s44342-024-00016-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
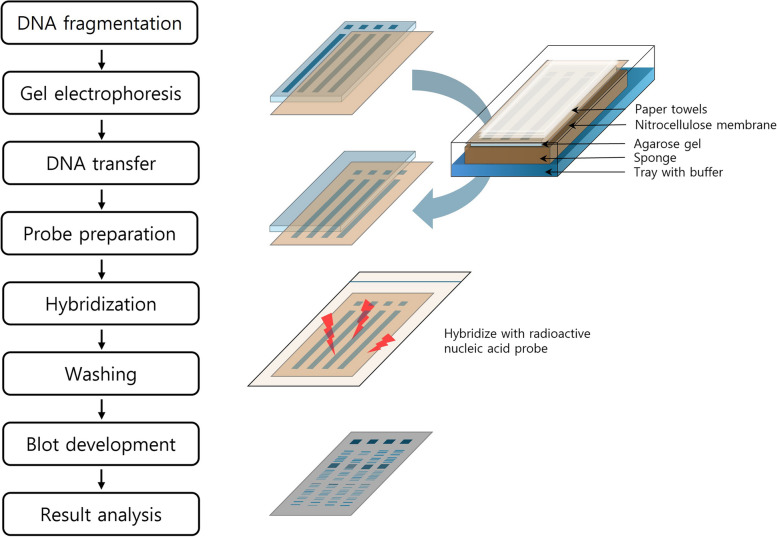
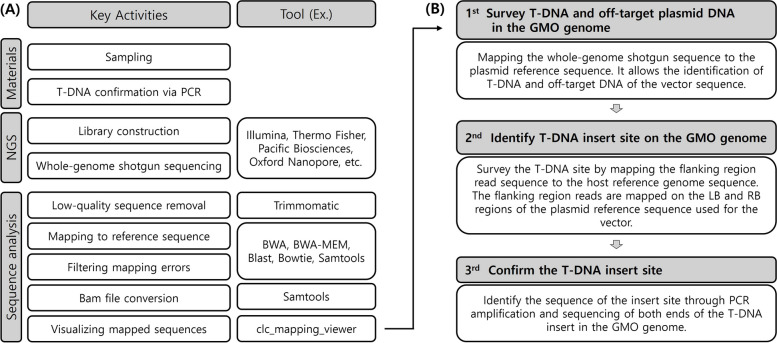
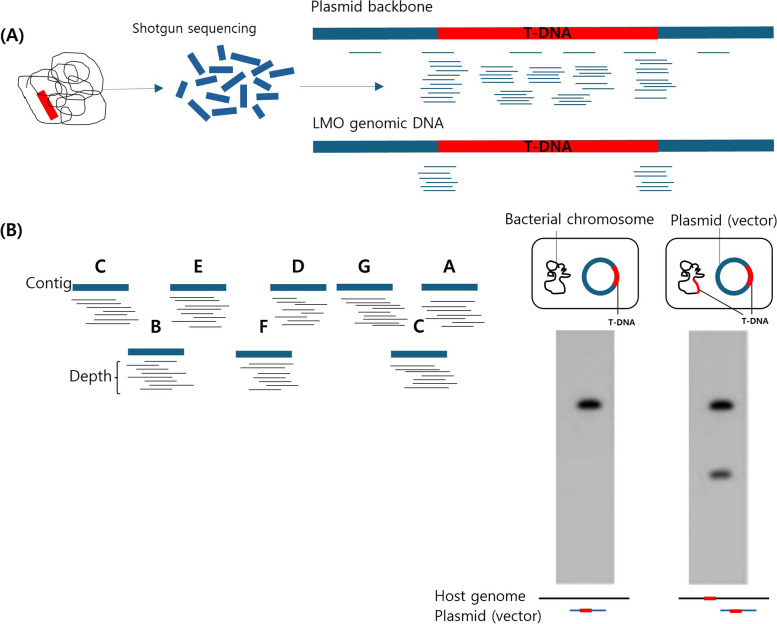
The molecular characterization of genetically modified organisms (GMOs) is essential for ensuring safety and gaining regulatory approval for commercialization. According to CODEX standards, this characterization involves evaluating the presence of introduced genes, insertion sites, copy number, and nucleotide sequence structure. Advances in technology have led to the increased use of next-generation sequencing (NGS) over traditional methods such as Southern blotting. While both methods provide high reproducibility and accuracy, Southern blotting is labor-intensive and time-consuming due to the need for repetitive probe design and analyses for each target, resulting in low throughput. Conversely, NGS facilitates rapid and comprehensive analysis by mapping whole-genome sequencing (WGS) data to plasmid sequences, accurately identifying T-DNA insertion sites and flanking regions. This advantage allows for efficient detection of T-DNA presence, copy number, and unintended gene insertions without additional probe work. This paper reviews the current status of GMO genome characterization using NGS and proposes more efficient strategies for this purpose.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
