Andrew M Stokely, Lane W Votapka, Marcus T Hock, Abigail E Teitgen, J Andrew McCammon, Andrew D McCulloch, Rommie E Amaro
{"title":"NetSci: A Library for High Performance Biomolecular Simulation Network Analysis Computation.","authors":"Andrew M Stokely, Lane W Votapka, Marcus T Hock, Abigail E Teitgen, J Andrew McCammon, Andrew D McCulloch, Rommie E Amaro","doi":"10.1021/acs.jcim.4c00899","DOIUrl":null,"url":null,"abstract":"<p><p>We present the NetSci program-an open-source scientific software package designed for estimating mutual information (MI) between data sets using GPU acceleration and a k-nearest-neighbor algorithm. This approach significantly enhances calculation speed, achieving improvements of several orders of magnitude over traditional CPU-based methods, with data set size limits dictated only by available hardware. To validate NetSci, we accurately compute MI for an analytically verifiable two-dimensional Gaussian distribution and replicate the generalized correlation (GC) analysis previously conducted on the B1 domain of protein G. We also apply NetSci to molecular dynamics simulations of the Sarcoendoplasmic Reticulum Calcium-ATPase (SERCA) pump, exploring the allosteric mechanisms and pathways influenced by ATP and 2'-deoxy-ATP (dATP) binding. Our analysis reveals distinct allosteric effects induced by ATP compared to dATP, with predicted information pathways from the bound nucleotide to the calcium-binding domain differing based on the nucleotide involved. NetSci proves to be a valuable tool for estimating MI and GC in various data sets and is particularly effective for analyzing intraprotein communication and information transfer.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"7966-7976"},"PeriodicalIF":5.3000,"publicationDate":"2024-10-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12039534/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00899","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
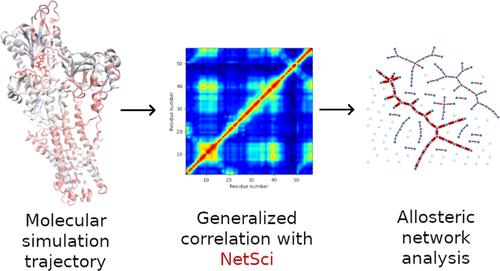
We present the NetSci program-an open-source scientific software package designed for estimating mutual information (MI) between data sets using GPU acceleration and a k-nearest-neighbor algorithm. This approach significantly enhances calculation speed, achieving improvements of several orders of magnitude over traditional CPU-based methods, with data set size limits dictated only by available hardware. To validate NetSci, we accurately compute MI for an analytically verifiable two-dimensional Gaussian distribution and replicate the generalized correlation (GC) analysis previously conducted on the B1 domain of protein G. We also apply NetSci to molecular dynamics simulations of the Sarcoendoplasmic Reticulum Calcium-ATPase (SERCA) pump, exploring the allosteric mechanisms and pathways influenced by ATP and 2'-deoxy-ATP (dATP) binding. Our analysis reveals distinct allosteric effects induced by ATP compared to dATP, with predicted information pathways from the bound nucleotide to the calcium-binding domain differing based on the nucleotide involved. NetSci proves to be a valuable tool for estimating MI and GC in various data sets and is particularly effective for analyzing intraprotein communication and information transfer.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
