Esraa A. Said , Ryan W. Lewis , Mark L. Dallas , Chris Peers , Fiona A. Ross , Asier Unciti-Broceta , D. Grahame Hardie , A. Mark Evans
{"title":"The thienopyridine A-769662 and benzimidazole 991 inhibit human TASK-3 potassium channels in an AMPK-independent manner","authors":"Esraa A. Said , Ryan W. Lewis , Mark L. Dallas , Chris Peers , Fiona A. Ross , Asier Unciti-Broceta , D. Grahame Hardie , A. Mark Evans","doi":"10.1016/j.bcp.2024.116562","DOIUrl":null,"url":null,"abstract":"<div><div>Heteromeric <u>T</u>andem pore domain <u>A</u>cid <u>S</u>ensitive (TASK)-1/3 channels are critical to oxygen-sensing by carotid body type 1 cells, where hypoxia-induced inhibition of TASK-3 and/or TASK-1/3 potassium currents leads to voltage-gated calcium entry, exocytotic transmitter release and increases in carotid body afferent input responses that initiate corrective changes in breathing patterns. It was proposed that, in response to hypoxia, the AMP–activated protein kinase (AMPK) might directly phosphorylate and inhibit TASK channels, in particular TASK–3, but studies on rat type I cells questioned this view. However, sequence alignment identified a putative AMPK recognition motif in human (h) TASK-3, but not hTASK–1, with Ser<sup>55</sup> representing a potential phosphorylation site. We therefore studied the effects of five different AMPK activators on recombinant hTASK–3 potassium channels expressed in human embryonic kidney (HEK)–293 cells. Two structurally unrelated AMPK activators, the thienopyridine A–769662 (100–500 µM) and the benzimidazole 991 (3–30 µM) inhibited hTASK–3 currents in a concentration–dependent manner, while the 4-azabenzimidazole MK–8722 (3–30 µM) partially inhibited hTASK–3 at concentrations above those required for maximal AMPK activation. By contrast, the 4-azabenzimidazole, BI-9774 (10–100 µM; a closely related analogue of MK8722) and the pro-drug AICA-riboside (1 mM; metabolised to ZMP, an AMP-mimetic) had no significant effect on hTASK–3 currents at concentrations sufficient to maximally activate AMPK. Importantly, A–769662 (300 µM) also inhibited hTASK–3 channel currents in HEK–293 cells that stably over-expressed an AMPK–β1 subunit mutant (S108A) that renders AMPK insensitive to activators that bind to the Allosteric Drug and Metabolite site, such as A–769662. We therefore identify A–769662 and 991 as novel hTASK–3 channel inhibitors and provide conclusive evidence that AMPK does not regulate hTASK–3 channel currents.</div></div>","PeriodicalId":8806,"journal":{"name":"Biochemical pharmacology","volume":"230 ","pages":"Article 116562"},"PeriodicalIF":5.6000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochemical pharmacology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0006295224005628","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
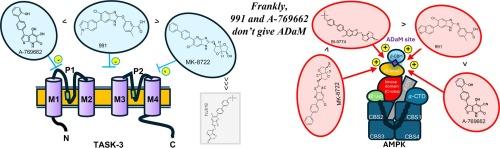
Heteromeric Tandem pore domain Acid Sensitive (TASK)-1/3 channels are critical to oxygen-sensing by carotid body type 1 cells, where hypoxia-induced inhibition of TASK-3 and/or TASK-1/3 potassium currents leads to voltage-gated calcium entry, exocytotic transmitter release and increases in carotid body afferent input responses that initiate corrective changes in breathing patterns. It was proposed that, in response to hypoxia, the AMP–activated protein kinase (AMPK) might directly phosphorylate and inhibit TASK channels, in particular TASK–3, but studies on rat type I cells questioned this view. However, sequence alignment identified a putative AMPK recognition motif in human (h) TASK-3, but not hTASK–1, with Ser55 representing a potential phosphorylation site. We therefore studied the effects of five different AMPK activators on recombinant hTASK–3 potassium channels expressed in human embryonic kidney (HEK)–293 cells. Two structurally unrelated AMPK activators, the thienopyridine A–769662 (100–500 µM) and the benzimidazole 991 (3–30 µM) inhibited hTASK–3 currents in a concentration–dependent manner, while the 4-azabenzimidazole MK–8722 (3–30 µM) partially inhibited hTASK–3 at concentrations above those required for maximal AMPK activation. By contrast, the 4-azabenzimidazole, BI-9774 (10–100 µM; a closely related analogue of MK8722) and the pro-drug AICA-riboside (1 mM; metabolised to ZMP, an AMP-mimetic) had no significant effect on hTASK–3 currents at concentrations sufficient to maximally activate AMPK. Importantly, A–769662 (300 µM) also inhibited hTASK–3 channel currents in HEK–293 cells that stably over-expressed an AMPK–β1 subunit mutant (S108A) that renders AMPK insensitive to activators that bind to the Allosteric Drug and Metabolite site, such as A–769662. We therefore identify A–769662 and 991 as novel hTASK–3 channel inhibitors and provide conclusive evidence that AMPK does not regulate hTASK–3 channel currents.
期刊介绍:
Biochemical Pharmacology publishes original research findings, Commentaries and review articles related to the elucidation of cellular and tissue function(s) at the biochemical and molecular levels, the modification of cellular phenotype(s) by genetic, transcriptional/translational or drug/compound-induced modifications, as well as the pharmacodynamics and pharmacokinetics of xenobiotics and drugs, the latter including both small molecules and biologics.
The journal''s target audience includes scientists engaged in the identification and study of the mechanisms of action of xenobiotics, biologics and drugs and in the drug discovery and development process.
All areas of cellular biology and cellular, tissue/organ and whole animal pharmacology fall within the scope of the journal. Drug classes covered include anti-infectives, anti-inflammatory agents, chemotherapeutics, cardiovascular, endocrinological, immunological, metabolic, neurological and psychiatric drugs, as well as research on drug metabolism and kinetics. While medicinal chemistry is a topic of complimentary interest, manuscripts in this area must contain sufficient biological data to characterize pharmacologically the compounds reported. Submissions describing work focused predominately on chemical synthesis and molecular modeling will not be considered for review.
While particular emphasis is placed on reporting the results of molecular and biochemical studies, research involving the use of tissue and animal models of human pathophysiology and toxicology is of interest to the extent that it helps define drug mechanisms of action, safety and efficacy.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
