{"title":"Modification of MM force fields around heme-Fe in the CYP–ligand complex and ab initio FMO calculations for the complex","authors":"Yoshinobu Nagura, Haruna Sabishiro, Nagomi Chimura, Masayuki Yuguchi, Narutoshi Tada, Daichi Takimoto, Noriyuki Kurita","doi":"10.1016/j.jmgm.2024.108875","DOIUrl":null,"url":null,"abstract":"<div><div>Cytochrome P450 (CYP) enzymes play essential roles in the synthesis and metabolic activation of physiologically active substances. CYP has a prosthetic heme (iron protoporphyrin IX) in its active center, where Fe ion (heme-Fe) is deeply involved in enzymatic reactions of CYP. To precisely describe the structure and electronic states around heme-Fe, we modified the force fields (FFs) around heme-Fe in molecular mechanics (MM) simulations and conducted <em>ab initio</em> fragment molecular orbital (FMO) calculations for the CYP–ligand complex. To describe the coordination bond between heme-Fe and its coordinated ligand (ketoconazole), we added FF between heme-Fe and the N atom of ketoconazole, and then the structure of the complex was optimized using the modified FF. Its adequacy was confirmed by comparing the MM-optimized structure with the X-ray crystal one of the CYP–ketoconazole complex. We also performed 100 ns molecular dynamics simulations and revealed that the coordination bonds around heme-Fe were maintained even at 310 K and that the CYP–ketoconazole structure was kept similar to the X-ray structure. Furthermore, we investigated the electronic states of the complex using the <em>ab initio</em> FMO method to identify the CYP residues and parts of ketoconazole that contribute to strong binding between CYP and ketoconazole. The present procedure of constructing FF between heme-Fe and ketoconazole can be applicable to other CYP–ligand complexes, and the modified FF can provide their accurate structures useful for predicting the specific interactions between CYP and its ligands.</div></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108875"},"PeriodicalIF":3.0000,"publicationDate":"2024-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S109332632400175X","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
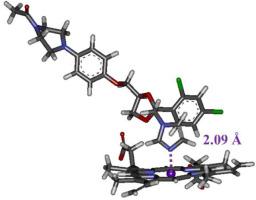
Cytochrome P450 (CYP) enzymes play essential roles in the synthesis and metabolic activation of physiologically active substances. CYP has a prosthetic heme (iron protoporphyrin IX) in its active center, where Fe ion (heme-Fe) is deeply involved in enzymatic reactions of CYP. To precisely describe the structure and electronic states around heme-Fe, we modified the force fields (FFs) around heme-Fe in molecular mechanics (MM) simulations and conducted ab initio fragment molecular orbital (FMO) calculations for the CYP–ligand complex. To describe the coordination bond between heme-Fe and its coordinated ligand (ketoconazole), we added FF between heme-Fe and the N atom of ketoconazole, and then the structure of the complex was optimized using the modified FF. Its adequacy was confirmed by comparing the MM-optimized structure with the X-ray crystal one of the CYP–ketoconazole complex. We also performed 100 ns molecular dynamics simulations and revealed that the coordination bonds around heme-Fe were maintained even at 310 K and that the CYP–ketoconazole structure was kept similar to the X-ray structure. Furthermore, we investigated the electronic states of the complex using the ab initio FMO method to identify the CYP residues and parts of ketoconazole that contribute to strong binding between CYP and ketoconazole. The present procedure of constructing FF between heme-Fe and ketoconazole can be applicable to other CYP–ligand complexes, and the modified FF can provide their accurate structures useful for predicting the specific interactions between CYP and its ligands.
修改 CYP 配体复合物中血红素-铁周围的 MM 力场,并对该复合物进行 ab initio FMO 计算。
细胞色素 P450(CYP)酶在生理活性物质的合成和代谢活化过程中发挥着重要作用。CYP 的活性中心有一个修复血红素(铁原卟啉 IX),其中的铁离子(血红素-铁)深度参与了 CYP 的酶促反应。为了精确描述血红素-铁周围的结构和电子状态,我们在分子力学(MM)模拟中修改了血红素-铁周围的力场(FFs),并对 CYP 配体复合物进行了 ab initio 片段分子轨道(FMO)计算。为了描述血红素-铁与其配位体(酮康唑)之间的配位键,我们在血红素-铁与酮康唑的 N 原子之间添加了 FF,然后使用修改后的 FF 对复合物结构进行了优化。通过将 MM 优化后的结构与 CYP-酮康唑复合物的 X 射线晶体结构进行比较,证实了 MM 结构的适当性。我们还进行了 100 ns 的分子动力学模拟,结果表明,即使在 310 K 的温度下,血红素-铁周围的配位键仍然保持不变,CYP-酮康唑的结构也与 X 射线结构相似。此外,我们还利用 ab initio FMO 方法研究了复合物的电子态,以确定 CYP 残基和酮康唑中有助于 CYP 与酮康唑强结合的部分。本研究构建血红素-铁和酮康唑之间的 FF 的过程可适用于其他 CYP 配体复合物,修饰后的 FF 可提供其精确结构,有助于预测 CYP 与其配体之间的特定相互作用。
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
