M. O. Mol, T. J. van Ham, N. Bannink, H. T. Bruggenwirth, J. C. Escher, J. M. Kros, J. J. M. Renkens, L. van Unen, R. M. Verdijk, J. Vlot, V. J. M. Verhoeven, S. Demirdas
{"title":"Biallelic and monoallelic variants in EFEMP1 can cause a severe and distinct subtype of heritable connective tissue disorder","authors":"M. O. Mol, T. J. van Ham, N. Bannink, H. T. Bruggenwirth, J. C. Escher, J. M. Kros, J. J. M. Renkens, L. van Unen, R. M. Verdijk, J. Vlot, V. J. M. Verhoeven, S. Demirdas","doi":"10.1038/s41431-024-01692-x","DOIUrl":null,"url":null,"abstract":"Variants in EFEMP1, encoding Fibulin-3, were previously reported as a rare cause of heritable connective tissue disorder (HCTD) with recurrent hernias and joint hypermobility. We report three new cases with biallelic or monoallelic EFEMP1 variants and severe hernia phenotypes. Two male siblings of 10 and 13 years old presented with marfanoid habitus, recurrent inguinal and umbilical hernias, generalized joint hypermobility, and scoliosis. Parents and halfsiblings reported joint hypermobility and umbilical hernias. The eldest boy died at age 16 from incarcerated gastrointestinal herniation complicated by gastric and bowel necrosis with perforation. Autopsy revealed widespread intestinal diverticula. Immunohistochemistry of skin and fascia tissue did not reveal any abnormalities, including normal staining of elastic fibers. Both siblings harbored compound heterozygous likely pathogenic EFEMP1 variants (c.1320 + 2T > A, p.? and c.698G > A, p.Gly233Asp). An unrelated 58-year-old male had marfanoid features, high myopia, recurrent diaphragmatic and inguinal hernias, and chronic gastrointestinal dilatation with severe malabsorption. Both his dizygotic twin-brother and mother had recurrent hernias and high myopia. This man died at 59 years of age, and autopsy showed extensive diaphragmatic herniation, bowel diverticula, and pulmonary emphysema. A heterozygous EFEMP1 splice-variant (c.81 + 1G > A, p.?) was identified, causing exon skipping leading to a start-loss. Targeted genome reanalysis nor RNA-sequencing revealed a second variant at the other allele. The reported individuals expand the clinical and pathological phenotypes of EFEMP1-related disease, a distinct entity within the spectrum of HCTD. The severe and recurrent hernias, gastrointestinal dilatation, and diverticulosis result in an increased risk for life-threatening complications, demanding early recognition and close monitoring.","PeriodicalId":12016,"journal":{"name":"European Journal of Human Genetics","volume":"32 12","pages":"1567-1573"},"PeriodicalIF":4.6000,"publicationDate":"2024-10-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s41431-024-01692-x.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41431-024-01692-x","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
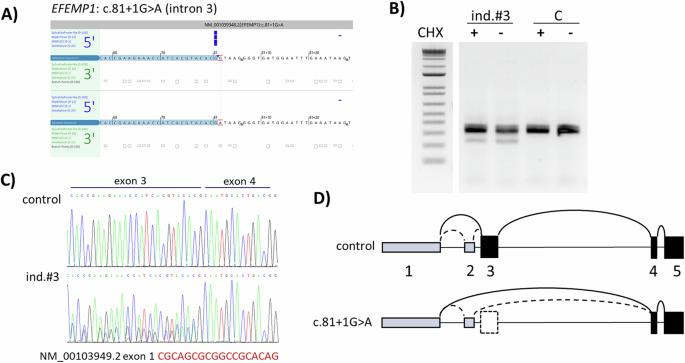
Variants in EFEMP1, encoding Fibulin-3, were previously reported as a rare cause of heritable connective tissue disorder (HCTD) with recurrent hernias and joint hypermobility. We report three new cases with biallelic or monoallelic EFEMP1 variants and severe hernia phenotypes. Two male siblings of 10 and 13 years old presented with marfanoid habitus, recurrent inguinal and umbilical hernias, generalized joint hypermobility, and scoliosis. Parents and halfsiblings reported joint hypermobility and umbilical hernias. The eldest boy died at age 16 from incarcerated gastrointestinal herniation complicated by gastric and bowel necrosis with perforation. Autopsy revealed widespread intestinal diverticula. Immunohistochemistry of skin and fascia tissue did not reveal any abnormalities, including normal staining of elastic fibers. Both siblings harbored compound heterozygous likely pathogenic EFEMP1 variants (c.1320 + 2T > A, p.? and c.698G > A, p.Gly233Asp). An unrelated 58-year-old male had marfanoid features, high myopia, recurrent diaphragmatic and inguinal hernias, and chronic gastrointestinal dilatation with severe malabsorption. Both his dizygotic twin-brother and mother had recurrent hernias and high myopia. This man died at 59 years of age, and autopsy showed extensive diaphragmatic herniation, bowel diverticula, and pulmonary emphysema. A heterozygous EFEMP1 splice-variant (c.81 + 1G > A, p.?) was identified, causing exon skipping leading to a start-loss. Targeted genome reanalysis nor RNA-sequencing revealed a second variant at the other allele. The reported individuals expand the clinical and pathological phenotypes of EFEMP1-related disease, a distinct entity within the spectrum of HCTD. The severe and recurrent hernias, gastrointestinal dilatation, and diverticulosis result in an increased risk for life-threatening complications, demanding early recognition and close monitoring.
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
