Luke W Silver, Elspeth A McLennan, Julian Beaman, Karen Burke da Silva, Peter Timms, Carolyn J Hogg, Katherine Belov
{"title":"Using bioinformatics to investigate functional diversity: a case study of MHC diversity in koalas.","authors":"Luke W Silver, Elspeth A McLennan, Julian Beaman, Karen Burke da Silva, Peter Timms, Carolyn J Hogg, Katherine Belov","doi":"10.1007/s00251-024-01356-6","DOIUrl":null,"url":null,"abstract":"<p><p>Conservation genomics can greatly improve conservation outcomes of threatened populations, including those impacted by disease. Understanding diversity within immune gene families, including the major histocompatibility complex (MHC) and toll-like receptors (TLR), is important due to the role they play in disease resilience and susceptibility. With recent advancements in sequencing technologies and bioinformatic tools, the cost of generating high-quality sequence data has significantly decreased and made it possible to investigate diversity across entire gene families in large numbers of individuals compared to investigating only a few genes or a few populations previously. Here, we use the koala as a case study for investigating functional diversity across populations. We utilised previous target enrichment data and 438 whole genomes to firstly, determine the level of sequencing depth required to investigate MHC diversity and, secondly, determine the current level of diversity in MHC genes in koala populations. We determined for low complexity, conserved genes such as TLR genes 10 × sequencing depth is sufficient to reliably genotype more than 90% of variants, whereas for complex genes such as the MHC greater than 20 × and preferably 30 × sequencing depth is required. We used whole genome data to identify 270 biallelic SNPs across 24 MHC genes as well as copy number variation (CNV) within class I and class II genes and conduct supertype analysis. Overall, we have provided a bioinformatic workflow for investigating variation in a complex immune gene family from whole genome sequencing data and determined current levels of diversity within koala MHC genes.</p>","PeriodicalId":13446,"journal":{"name":"Immunogenetics","volume":" ","pages":"381-395"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11496358/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00251-024-01356-6","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/5 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
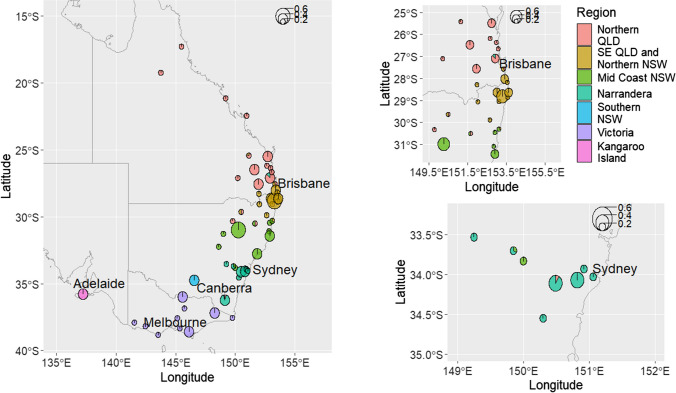
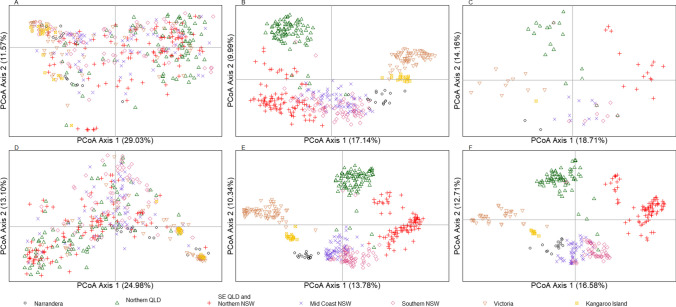
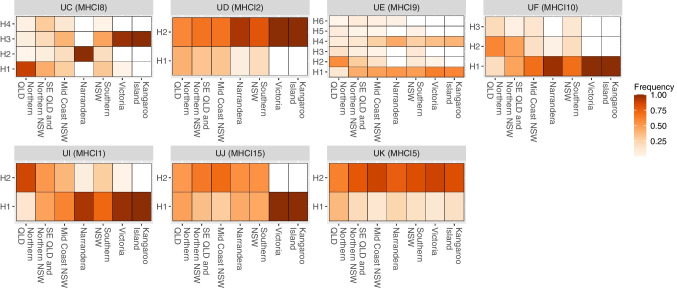
Conservation genomics can greatly improve conservation outcomes of threatened populations, including those impacted by disease. Understanding diversity within immune gene families, including the major histocompatibility complex (MHC) and toll-like receptors (TLR), is important due to the role they play in disease resilience and susceptibility. With recent advancements in sequencing technologies and bioinformatic tools, the cost of generating high-quality sequence data has significantly decreased and made it possible to investigate diversity across entire gene families in large numbers of individuals compared to investigating only a few genes or a few populations previously. Here, we use the koala as a case study for investigating functional diversity across populations. We utilised previous target enrichment data and 438 whole genomes to firstly, determine the level of sequencing depth required to investigate MHC diversity and, secondly, determine the current level of diversity in MHC genes in koala populations. We determined for low complexity, conserved genes such as TLR genes 10 × sequencing depth is sufficient to reliably genotype more than 90% of variants, whereas for complex genes such as the MHC greater than 20 × and preferably 30 × sequencing depth is required. We used whole genome data to identify 270 biallelic SNPs across 24 MHC genes as well as copy number variation (CNV) within class I and class II genes and conduct supertype analysis. Overall, we have provided a bioinformatic workflow for investigating variation in a complex immune gene family from whole genome sequencing data and determined current levels of diversity within koala MHC genes.
期刊介绍:
Immunogenetics publishes original papers, brief communications, and reviews on research in the following areas: genetics and evolution of the immune system; genetic control of immune response and disease susceptibility; bioinformatics of the immune system; structure of immunologically important molecules; and immunogenetics of reproductive biology, tissue differentiation, and development.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
