Ana Molina-Taborda, Pilar Cossio, Olga Lopez-Acevedo, Marylou Gabrié
{"title":"Active Learning of Boltzmann Samplers and Potential Energies with Quantum Mechanical Accuracy.","authors":"Ana Molina-Taborda, Pilar Cossio, Olga Lopez-Acevedo, Marylou Gabrié","doi":"10.1021/acs.jctc.4c00506","DOIUrl":null,"url":null,"abstract":"<p><p>Extracting consistent statistics between relevant free energy minima of a molecular system is essential for physics, chemistry, and biology. Molecular dynamics (MD) simulations can aid in this task but are computationally expensive, especially for systems that require quantum accuracy. To overcome this challenge, we developed an approach combining enhanced sampling with deep generative models and active learning of a machine learning potential (MLP). We introduce an adaptive Markov chain Monte Carlo framework that enables the training of one normalizing flow (NF) and one MLP per state, achieving rapid convergence toward the Boltzmann distribution. Leveraging the trained NF and MLP models, we compute thermodynamic observables such as free energy differences and optical spectra. We apply this method to study the isomerization of an ultrasmall silver nanocluster belonging to a set of systems with diverse applications in the fields of medicine and catalysis.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"8833-8843"},"PeriodicalIF":5.5000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00506","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/6 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
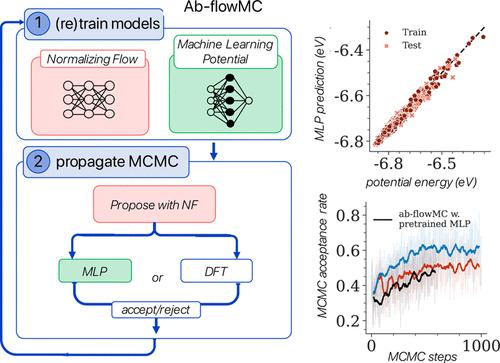
Extracting consistent statistics between relevant free energy minima of a molecular system is essential for physics, chemistry, and biology. Molecular dynamics (MD) simulations can aid in this task but are computationally expensive, especially for systems that require quantum accuracy. To overcome this challenge, we developed an approach combining enhanced sampling with deep generative models and active learning of a machine learning potential (MLP). We introduce an adaptive Markov chain Monte Carlo framework that enables the training of one normalizing flow (NF) and one MLP per state, achieving rapid convergence toward the Boltzmann distribution. Leveraging the trained NF and MLP models, we compute thermodynamic observables such as free energy differences and optical spectra. We apply this method to study the isomerization of an ultrasmall silver nanocluster belonging to a set of systems with diverse applications in the fields of medicine and catalysis.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
