{"title":"A clinically feasible algorithm for the parallel detection of glioma-associated copy number variation markers based on shallow whole genome sequencing","authors":"Shuai Wu, Chenyu Ma, Jiawei Cai, Chenkang Yang, Xiaojia Liu, Chen Luo, Jingyi Yang, Zhang Xiong, Dandan Cao, Hong Chen","doi":"10.1002/2056-4538.70005","DOIUrl":null,"url":null,"abstract":"<p>Molecular features are incorporated into the integrated diagnostic system for adult diffuse gliomas. Of these, copy number variation (CNV) markers, including both arm-level (1p/19q codeletion, +7/−10 signature) and gene-level (<i>EGFR</i> gene amplification, <i>CDKN2A/B</i> homozygous deletion) changes, have revolutionized the diagnostic paradigm by updating the subtyping and grading schemes. Shallow whole genome sequencing (sWGS) has been widely used for CNV detection due to its cost-effectiveness and versatility. However, the parallel detection of glioma-associated CNV markers using sWGS has not been optimized in a clinical setting. Herein, we established a model-based approach to classify the CNV status of glioma-associated diagnostic markers with a single test. To enhance its clinical utility, we carried out hypothesis testing model-based analysis through the estimation of copy ratio fluctuation level, which was implemented individually and independently and, thus, avoided the necessity for normal controls. Besides, the customization of required minimal tumor fraction (TF) was evaluated and recommended for each glioma-associated marker to ensure robust classification. As a result, with 1× sequencing depth and 0.05 TF, arm-level CNVs could be reliably detected with at least 99.5% sensitivity and specificity. For <i>EGFR</i> gene amplification and <i>CDKN2A/B</i> homozygous deletion, the corresponding TF limits were 0.15 and 0.45 to ensure the evaluation metrics were both higher than 97%. Furthermore, we applied the algorithm to an independent glioma cohort and observed the expected sample distribution and prognostic stratification patterns. In conclusion, we provide a clinically applicable algorithm to classify the CNV status of glioma-associated markers in parallel.</p>","PeriodicalId":48612,"journal":{"name":"Journal of Pathology Clinical Research","volume":"10 6","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2024-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11458885/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Pathology Clinical Research","FirstCategoryId":"3","ListUrlMain":"https://pathsocjournals.onlinelibrary.wiley.com/doi/10.1002/2056-4538.70005","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PATHOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
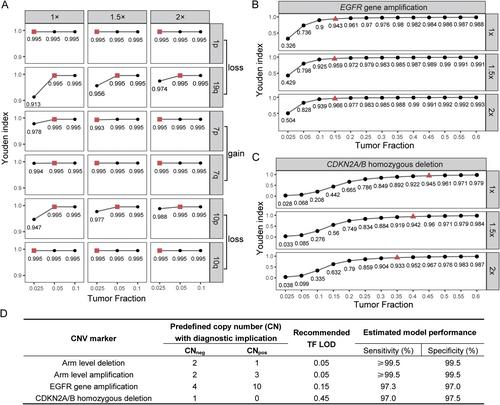
Molecular features are incorporated into the integrated diagnostic system for adult diffuse gliomas. Of these, copy number variation (CNV) markers, including both arm-level (1p/19q codeletion, +7/−10 signature) and gene-level (EGFR gene amplification, CDKN2A/B homozygous deletion) changes, have revolutionized the diagnostic paradigm by updating the subtyping and grading schemes. Shallow whole genome sequencing (sWGS) has been widely used for CNV detection due to its cost-effectiveness and versatility. However, the parallel detection of glioma-associated CNV markers using sWGS has not been optimized in a clinical setting. Herein, we established a model-based approach to classify the CNV status of glioma-associated diagnostic markers with a single test. To enhance its clinical utility, we carried out hypothesis testing model-based analysis through the estimation of copy ratio fluctuation level, which was implemented individually and independently and, thus, avoided the necessity for normal controls. Besides, the customization of required minimal tumor fraction (TF) was evaluated and recommended for each glioma-associated marker to ensure robust classification. As a result, with 1× sequencing depth and 0.05 TF, arm-level CNVs could be reliably detected with at least 99.5% sensitivity and specificity. For EGFR gene amplification and CDKN2A/B homozygous deletion, the corresponding TF limits were 0.15 and 0.45 to ensure the evaluation metrics were both higher than 97%. Furthermore, we applied the algorithm to an independent glioma cohort and observed the expected sample distribution and prognostic stratification patterns. In conclusion, we provide a clinically applicable algorithm to classify the CNV status of glioma-associated markers in parallel.
期刊介绍:
The Journal of Pathology: Clinical Research and The Journal of Pathology serve as translational bridges between basic biomedical science and clinical medicine with particular emphasis on, but not restricted to, tissue based studies.
The focus of The Journal of Pathology: Clinical Research is the publication of studies that illuminate the clinical relevance of research in the broad area of the study of disease. Appropriately powered and validated studies with novel diagnostic, prognostic and predictive significance, and biomarker discover and validation, will be welcomed. Studies with a predominantly mechanistic basis will be more appropriate for the companion Journal of Pathology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
