{"title":"GLTSCR1 deficiency promotes colorectal cancer development through regulating non-homologous end joining","authors":"Fengyan Han, Xiaoxu Zhou, Lu Liu, Beibei Yang, Pengyuan Liu, Enping Xu, Zhe Tang, Honghe Zhang","doi":"10.1038/s41388-024-03179-x","DOIUrl":null,"url":null,"abstract":"Non-homologous end joining (NHEJ), as one major pathway of DNA double-strand break (DSB) repair, could cause genomic instability, which plays pivotal roles in cancer development. While, chromatin remodeling complexes dictate the selection and orchestration of DSB repair pathways by regulating chromatin dynamics. However, the crosstalk between NHEJ and chromatin remodeling in cancer progress remains unclear. In this study, deficiency of GLTSCR1 causes resistance to DNA damage in colorectal cancer (CRC) cells by promoting NHEJ repair efficiency. Mechanistically, GLTSCR1 interacts with BRD9 to engage in the assembly of the non-canonical BAF complex (GBAF). However, GLTSCR1 deficiency disrupts GBAF and triggers the ubiquitination degradation of BRD9. Furthermore, GLTSCR1 deficiency causes aberrant opening in the promoter region of NHEJ repair-associated genes, which promotes CRC development. While, GLTSCR1 and its binding partner BRD9 are not directly involved in assembling NHEJ repair machinery; instead, they regulate the DNA accessibility of NHEJ repair-associated genes. Collectively, our findings confirm GLTSCR1 deficiency as a critical regulatory event of the NHEJ pathway in CRC development, which might require different therapeutic strategy for GLTSCR1 wild-type and mutant CRC.","PeriodicalId":19524,"journal":{"name":"Oncogene","volume":"43 48","pages":"3517-3531"},"PeriodicalIF":7.3000,"publicationDate":"2024-10-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Oncogene","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s41388-024-03179-x","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
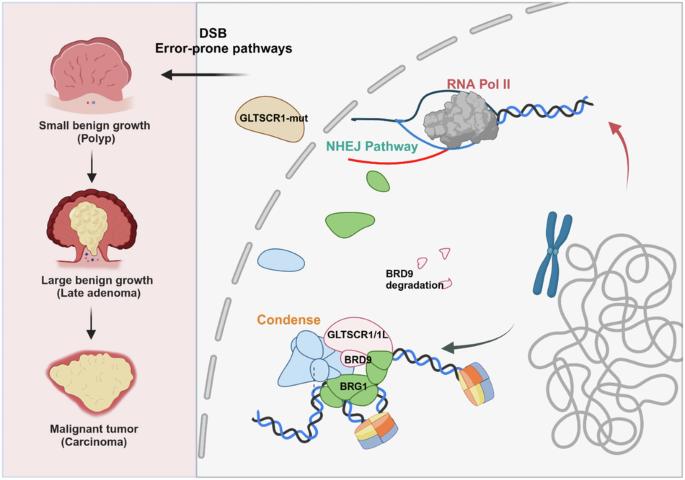
Non-homologous end joining (NHEJ), as one major pathway of DNA double-strand break (DSB) repair, could cause genomic instability, which plays pivotal roles in cancer development. While, chromatin remodeling complexes dictate the selection and orchestration of DSB repair pathways by regulating chromatin dynamics. However, the crosstalk between NHEJ and chromatin remodeling in cancer progress remains unclear. In this study, deficiency of GLTSCR1 causes resistance to DNA damage in colorectal cancer (CRC) cells by promoting NHEJ repair efficiency. Mechanistically, GLTSCR1 interacts with BRD9 to engage in the assembly of the non-canonical BAF complex (GBAF). However, GLTSCR1 deficiency disrupts GBAF and triggers the ubiquitination degradation of BRD9. Furthermore, GLTSCR1 deficiency causes aberrant opening in the promoter region of NHEJ repair-associated genes, which promotes CRC development. While, GLTSCR1 and its binding partner BRD9 are not directly involved in assembling NHEJ repair machinery; instead, they regulate the DNA accessibility of NHEJ repair-associated genes. Collectively, our findings confirm GLTSCR1 deficiency as a critical regulatory event of the NHEJ pathway in CRC development, which might require different therapeutic strategy for GLTSCR1 wild-type and mutant CRC.
期刊介绍:
Oncogene is dedicated to advancing our understanding of cancer processes through the publication of exceptional research. The journal seeks to disseminate work that challenges conventional theories and contributes to establishing new paradigms in the etio-pathogenesis, diagnosis, treatment, or prevention of cancers. Emphasis is placed on research shedding light on processes driving metastatic spread and providing crucial insights into cancer biology beyond existing knowledge.
Areas covered include the cellular and molecular biology of cancer, resistance to cancer therapies, and the development of improved approaches to enhance survival. Oncogene spans the spectrum of cancer biology, from fundamental and theoretical work to translational, applied, and clinical research, including early and late Phase clinical trials, particularly those with biologic and translational endpoints.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
