{"title":"An Open-Source Implementation of the Scaffold Identification and Naming System (SCINS) and Example Applications","authors":"Kamen P. Petrov, Andreas Bender","doi":"10.1021/acs.jcim.4c01314","DOIUrl":null,"url":null,"abstract":"Organizing and partitioning sets of chemical structures is of considerable practical significance, e.g., in compound library analysis and the postprocessing of screening hit lists. Approaches such as unsupervised clustering are computationally demanding and dataset-dependent; on the other hand, rule-based methods, such as those based on Murcko scaffolds, have linear time complexity but are often too fine-grained, leading to a large number of singletons or sparsely populated classes. An alternative rule-based method that seeks to achieve an optimal balance when grouping compounds into sets is the ‘Scaffold Identification and Naming System’ (SCINS). To facilitate public use of this previously published method, here, we provide an open-source Python implementation of SCINS, dependent only on RDKit. We show that SCINS can be useful in identifying sparsely and densely populated regions in chemical space in large databases, here exemplified with Enamine REAL Diverse and ChEMBL. We find that Enamine REAL Diverse covers a much smaller SCINS space relative to ChEMBL, whereas the opposite is true when Murcko and generic Murcko scaffolds are considered. Additionally, we show that SCINS can result in chemically intuitive grouping of medium-sized sets of bioactive compounds, which can be useful in compound selection from virtual screening campaigns as well as postprocessing of experimental hit lists. Hence, in this work, we provide both an open-source implementation of SCINS and its characterization with relevant use cases.","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"86 1","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2024-10-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c01314","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
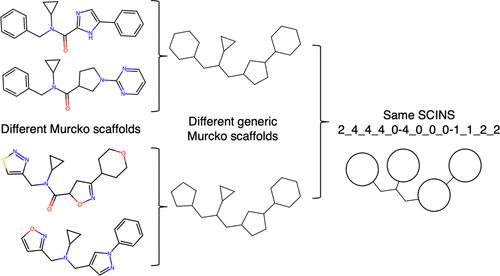
Organizing and partitioning sets of chemical structures is of considerable practical significance, e.g., in compound library analysis and the postprocessing of screening hit lists. Approaches such as unsupervised clustering are computationally demanding and dataset-dependent; on the other hand, rule-based methods, such as those based on Murcko scaffolds, have linear time complexity but are often too fine-grained, leading to a large number of singletons or sparsely populated classes. An alternative rule-based method that seeks to achieve an optimal balance when grouping compounds into sets is the ‘Scaffold Identification and Naming System’ (SCINS). To facilitate public use of this previously published method, here, we provide an open-source Python implementation of SCINS, dependent only on RDKit. We show that SCINS can be useful in identifying sparsely and densely populated regions in chemical space in large databases, here exemplified with Enamine REAL Diverse and ChEMBL. We find that Enamine REAL Diverse covers a much smaller SCINS space relative to ChEMBL, whereas the opposite is true when Murcko and generic Murcko scaffolds are considered. Additionally, we show that SCINS can result in chemically intuitive grouping of medium-sized sets of bioactive compounds, which can be useful in compound selection from virtual screening campaigns as well as postprocessing of experimental hit lists. Hence, in this work, we provide both an open-source implementation of SCINS and its characterization with relevant use cases.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
