Discovery of quinazoline-benzothiazole derivatives as novel potent protease-activated receptor 4 antagonists with improved pharmacokinetics and low bleeding liability
Shanshan Li , Shangde Liu , Duo Yuan , Renjie Liu , Lifang Hu , Xiong Zhu
{"title":"Discovery of quinazoline-benzothiazole derivatives as novel potent protease-activated receptor 4 antagonists with improved pharmacokinetics and low bleeding liability","authors":"Shanshan Li , Shangde Liu , Duo Yuan , Renjie Liu , Lifang Hu , Xiong Zhu","doi":"10.1016/j.ejmech.2024.116980","DOIUrl":null,"url":null,"abstract":"<div><div>Protease-activated receptor 4 (PAR4) plays a critical role in the development of pathological thrombosis, and targeting PAR4 is considered a promising strategy for improving antiplatelet therapies. Here, we reported the design of a series of quinazoline-benzothiazole-based PAR4 antagonists using a scaffold-hopping strategy. Systematic structure-activity relationship exploration leads to the discovery of compounds <strong>20f</strong> and 2<strong>0g</strong>, which displayed optimal activity (<em>h</em>. PAR4-AP PRP IC<sub>50</sub> = 6.39 nM and 3.45 nM, respectively) on human platelets and high selectivity for PAR4. Both of them also showed excellent metabolic stability in human liver microsomes (compound <strong>20f</strong>, T<sub>1/2</sub> = 249.83 min, compound <strong>20g</strong>, T<sub>1/2</sub> = 282.60 min) and favourable PK profiles in rats (compound <strong>20f</strong>, T<sub>1/2</sub> = 5.16 h, <em>F</em> = 50.5 %, compound <strong>20g</strong>, T<sub>1/2</sub> = 7.05 h, <em>F</em> = 27.3 %). More importantly, neither compound prolonged the bleeding time in the mouse tail-cutting model (10 mg/kg, <em>p.o</em>.). These results suggest that these compounds have great potential for use in antiplatelet therapies.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"280 ","pages":"Article 116980"},"PeriodicalIF":5.9000,"publicationDate":"2024-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0223523424008626","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
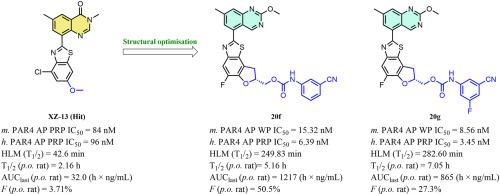
Protease-activated receptor 4 (PAR4) plays a critical role in the development of pathological thrombosis, and targeting PAR4 is considered a promising strategy for improving antiplatelet therapies. Here, we reported the design of a series of quinazoline-benzothiazole-based PAR4 antagonists using a scaffold-hopping strategy. Systematic structure-activity relationship exploration leads to the discovery of compounds 20f and 20g, which displayed optimal activity (h. PAR4-AP PRP IC50 = 6.39 nM and 3.45 nM, respectively) on human platelets and high selectivity for PAR4. Both of them also showed excellent metabolic stability in human liver microsomes (compound 20f, T1/2 = 249.83 min, compound 20g, T1/2 = 282.60 min) and favourable PK profiles in rats (compound 20f, T1/2 = 5.16 h, F = 50.5 %, compound 20g, T1/2 = 7.05 h, F = 27.3 %). More importantly, neither compound prolonged the bleeding time in the mouse tail-cutting model (10 mg/kg, p.o.). These results suggest that these compounds have great potential for use in antiplatelet therapies.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
