Electronic and geometric features controlling the reactivity of Mg-vanadate and V2O5 surfaces toward the initial C–H activation of C1–C3 alkanes – A DFT+U study
Hansel Montalvo-Castro , Álvaro Loaiza-Orduz , Randall J. Meyer , Craig Plaisance , David Hibbitts
{"title":"Electronic and geometric features controlling the reactivity of Mg-vanadate and V2O5 surfaces toward the initial C–H activation of C1–C3 alkanes – A DFT+U study","authors":"Hansel Montalvo-Castro , Álvaro Loaiza-Orduz , Randall J. Meyer , Craig Plaisance , David Hibbitts","doi":"10.1016/j.jcat.2024.115800","DOIUrl":null,"url":null,"abstract":"<div><div>This work employs density functional theory (DFT+U) calculations to explore initial C–H activations in C<sub>1</sub>–C<sub>3</sub> alkanes on V<sub>2</sub>O<sub>5</sub>, MgV<sub>2</sub>O<sub>6</sub> (meta-vanadate), Mg<sub>2</sub>V<sub>2</sub>O<sub>7</sub> (pyro-vanadate), and Mg<sub>3</sub>V<sub>2</sub>O<sub>8</sub> (ortho-vanadate) surfaces. These materials are selective catalysts for the oxidative dehydrogenation (ODH) of alkanes into alkenes, which offers practical and thermodynamic advantages over non-oxidative alkane dehydrogenation. The geometric and electronic properties that govern the reactivity of these materials, however, have not been explored by theory despite their importance in controlling rate determining alkane initial C–H activation during ODH catalysis. In this work, we explore fourteen low-energy surfaces of Mg<em><sub>x</sub></em>V<sub>2</sub>O<em><sub>x</sub></em><sub>+5</sub> (<em>x</em> = 0–3) exposing 64 distinct O atoms (reaction sites). C–H activation barriers are largest on Mg<sub>3</sub>V<sub>2</sub>O<sub>8</sub>, lower and similar for Mg<sub>2</sub>V<sub>2</sub>O<sub>7</sub> and MgV<sub>2</sub>O<sub>6</sub>, and lowest for V<sub>2</sub>O<sub>5</sub> surfaces; these predicted trends are consistent with measured ODH reactivity in earlier studies. Barriers are lowest (on average) when alkanes react with O atoms bound to a single V atom, with bridging O atoms having slightly higher barriers, and three-fold O atoms having the largest activation barriers. However, there is scattering within each subset indicating that factors beyond O-atom coordination have a significant role in the barriers. Vacancy formation energies (VFE) and the O 2p band energies were found to be weak descriptors of surface O reactivity for alkane activation barriers. Hydrogen addition energy (HAE) and methyl addition energy (MAE) values, in contrast, were found to correlate well with alkane activation barriers. MAE, however, outperforms HAE correlations because of the tendency of H* to form H-bonds with nearby surface O atoms, and those H-bonds are absent in C–H activation transition states causing scatter in the correlation of barriers with HAE. Constrained-orbital DFT methods were used to establish a theoretical thermochemical cycle that decouples surface reduction by CH<sub>3</sub>* into three components: surface distortion, orbital localization, and bond formation. These results give insights into how Mg:V ratios, surface structure (O-atom coordination), and reducibility (HAE, MAE) impact the reactivity of vanadium-based metal oxides toward alkane activation.</div></div>","PeriodicalId":346,"journal":{"name":"Journal of Catalysis","volume":"442 ","pages":"Article 115800"},"PeriodicalIF":6.5000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Catalysis","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S002195172400513X","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/18 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
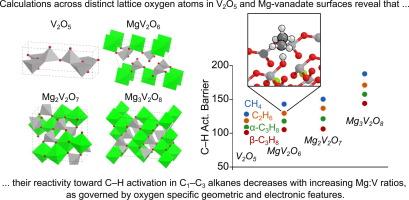
This work employs density functional theory (DFT+U) calculations to explore initial C–H activations in C1–C3 alkanes on V2O5, MgV2O6 (meta-vanadate), Mg2V2O7 (pyro-vanadate), and Mg3V2O8 (ortho-vanadate) surfaces. These materials are selective catalysts for the oxidative dehydrogenation (ODH) of alkanes into alkenes, which offers practical and thermodynamic advantages over non-oxidative alkane dehydrogenation. The geometric and electronic properties that govern the reactivity of these materials, however, have not been explored by theory despite their importance in controlling rate determining alkane initial C–H activation during ODH catalysis. In this work, we explore fourteen low-energy surfaces of MgxV2Ox+5 (x = 0–3) exposing 64 distinct O atoms (reaction sites). C–H activation barriers are largest on Mg3V2O8, lower and similar for Mg2V2O7 and MgV2O6, and lowest for V2O5 surfaces; these predicted trends are consistent with measured ODH reactivity in earlier studies. Barriers are lowest (on average) when alkanes react with O atoms bound to a single V atom, with bridging O atoms having slightly higher barriers, and three-fold O atoms having the largest activation barriers. However, there is scattering within each subset indicating that factors beyond O-atom coordination have a significant role in the barriers. Vacancy formation energies (VFE) and the O 2p band energies were found to be weak descriptors of surface O reactivity for alkane activation barriers. Hydrogen addition energy (HAE) and methyl addition energy (MAE) values, in contrast, were found to correlate well with alkane activation barriers. MAE, however, outperforms HAE correlations because of the tendency of H* to form H-bonds with nearby surface O atoms, and those H-bonds are absent in C–H activation transition states causing scatter in the correlation of barriers with HAE. Constrained-orbital DFT methods were used to establish a theoretical thermochemical cycle that decouples surface reduction by CH3* into three components: surface distortion, orbital localization, and bond formation. These results give insights into how Mg:V ratios, surface structure (O-atom coordination), and reducibility (HAE, MAE) impact the reactivity of vanadium-based metal oxides toward alkane activation.
期刊介绍:
The Journal of Catalysis publishes scholarly articles on both heterogeneous and homogeneous catalysis, covering a wide range of chemical transformations. These include various types of catalysis, such as those mediated by photons, plasmons, and electrons. The focus of the studies is to understand the relationship between catalytic function and the underlying chemical properties of surfaces and metal complexes.
The articles in the journal offer innovative concepts and explore the synthesis and kinetics of inorganic solids and homogeneous complexes. Furthermore, they discuss spectroscopic techniques for characterizing catalysts, investigate the interaction of probes and reacting species with catalysts, and employ theoretical methods.
The research presented in the journal should have direct relevance to the field of catalytic processes, addressing either fundamental aspects or applications of catalysis.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
