Ekin Esme Bas, Karen Marlenne Garcia Alvarez, Andreas Schneemann, Thomas Heine, Dorothea Golze
{"title":"Robust Computation and Analysis of Vibrational Spectra of Layered Framework Materials Including Host–Guest Interactions","authors":"Ekin Esme Bas, Karen Marlenne Garcia Alvarez, Andreas Schneemann, Thomas Heine, Dorothea Golze","doi":"10.1021/acs.jctc.4c01021","DOIUrl":null,"url":null,"abstract":"Layered framework materials, a rapidly advancing class of porous materials, are composed of molecular components stitched together via covalent bonds and are usually synthesized through wet-chemical methods. Computational infrared (IR) and Raman spectra are among the most important characterization tools for this material class. Besides the <i>a priori</i> known spectra of the molecular building blocks and the solvent, they allow for <i>in situ</i> monitoring of the framework formation during synthesis. Therefore, they need to capture the additional peaks from host–guest interactions and the bands from emerging bonds between the molecular building blocks, verifying the successful synthesis of the desired material. In this work, we propose a robust computational framework based on <i>ab initio</i> molecular dynamics (AIMD), where we compute IR and Raman spectra from the time-correlation functions of dipole moments and polarizability tensors, respectively. As a case study, we apply our methodology to a covalent organic framework (COF) material, COF-1, and present its AIMD-computed IR and Raman spectra with and without 1,4-dioxane solvent molecules in its pores. To determine robust settings, we meticulously validate our model and explore how stacking disorder and different methods for computing dipole moments and polarizabilities affect IR and Raman intensities. Using our robust computational protocol, we achieve excellent agreement with experimental data. Furthermore, we illustrate how the computed spectra can be dissected into individual contributions from the solvent molecules, the molecular building blocks of COF-1, and the bonds connecting them.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"75 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-10-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01021","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
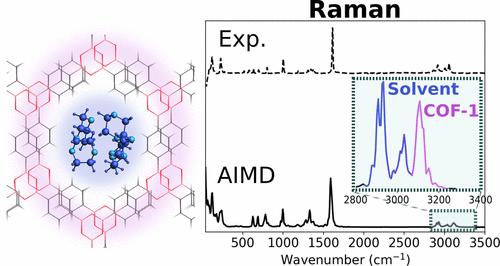
Layered framework materials, a rapidly advancing class of porous materials, are composed of molecular components stitched together via covalent bonds and are usually synthesized through wet-chemical methods. Computational infrared (IR) and Raman spectra are among the most important characterization tools for this material class. Besides the a priori known spectra of the molecular building blocks and the solvent, they allow for in situ monitoring of the framework formation during synthesis. Therefore, they need to capture the additional peaks from host–guest interactions and the bands from emerging bonds between the molecular building blocks, verifying the successful synthesis of the desired material. In this work, we propose a robust computational framework based on ab initio molecular dynamics (AIMD), where we compute IR and Raman spectra from the time-correlation functions of dipole moments and polarizability tensors, respectively. As a case study, we apply our methodology to a covalent organic framework (COF) material, COF-1, and present its AIMD-computed IR and Raman spectra with and without 1,4-dioxane solvent molecules in its pores. To determine robust settings, we meticulously validate our model and explore how stacking disorder and different methods for computing dipole moments and polarizabilities affect IR and Raman intensities. Using our robust computational protocol, we achieve excellent agreement with experimental data. Furthermore, we illustrate how the computed spectra can be dissected into individual contributions from the solvent molecules, the molecular building blocks of COF-1, and the bonds connecting them.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
