{"title":"Dirac States versus Nearly-Free-Electron States in Ternary C2As6(1–x)P6x Monolayers – A Density Functional Theory Study","authors":"Amrendra Kumar, C. Kamal","doi":"10.1021/acs.jpcc.4c04414","DOIUrl":null,"url":null,"abstract":"Using density functional theory-based calculations, we study the stability, geometric, and electronic properties of ternary C<sub>2</sub>As<sub>6(1–<i>x</i>)</sub>P<sub>6<i>x</i></sub> monolayers in a buckled honeycomb lattice. As the composition of P atoms (<i>x</i>) varies from 0 to 1 in steps of 1/6, we generated a total of 13 different possible geometric configurations. Binding energy calculations and <i>ab initio</i> molecular dynamics simulations under ambient conditions predict the stability of the ternary monolayers. Their electronic band structure and density of states calculations reveal a rich variety of electronic properties, ranging from semimetal to semiconducting or metallic, depending on the value of <i>x</i> and the crystal symmetry. The ternary C<sub>2</sub>As<sub>6(1–<i>x</i>)</sub>P<sub>6<i>x</i></sub> monolayers possess two distinct electronic dispersions near the Fermi level: a linear dispersion (Dirac cone) around the high-symmetry <i>K</i> point and a nearly-free-electron (NFE)-like parabolic dispersion around the high-symmetry Γ point in the Brillouin zone. Isoelectronic substitution breaks some spatial symmetries, causing the Dirac cones to become anisotropic and unpinned from the <i>K</i> point. It also leads to the opening of a small energy gap between the Dirac cone states. In contrast, the nature of the NFE-like parabolic dispersion remains nearly unaltered by the crystal symmetry. Notably, the minimum energy of the NFE band and the effective mass of the electron can be tuned by the composition of the ternary monolayers. Detailed orbital analysis reveals that the contributions of <i>s</i> and <i>p</i><sub><i>z</i></sub> orbitals of C atoms play a crucial role in tuning the NFE states. Overall, we reveal the novel and versatile electronic properties of the ternary monolayers and demonstrate their tunability via composition.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"112 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c04414","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
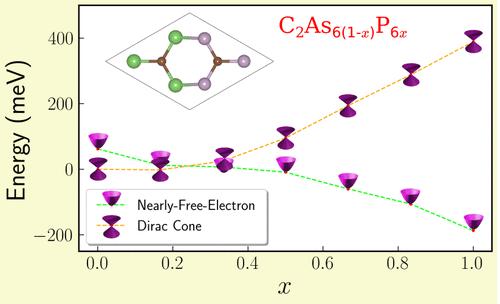
Using density functional theory-based calculations, we study the stability, geometric, and electronic properties of ternary C2As6(1–x)P6x monolayers in a buckled honeycomb lattice. As the composition of P atoms (x) varies from 0 to 1 in steps of 1/6, we generated a total of 13 different possible geometric configurations. Binding energy calculations and ab initio molecular dynamics simulations under ambient conditions predict the stability of the ternary monolayers. Their electronic band structure and density of states calculations reveal a rich variety of electronic properties, ranging from semimetal to semiconducting or metallic, depending on the value of x and the crystal symmetry. The ternary C2As6(1–x)P6x monolayers possess two distinct electronic dispersions near the Fermi level: a linear dispersion (Dirac cone) around the high-symmetry K point and a nearly-free-electron (NFE)-like parabolic dispersion around the high-symmetry Γ point in the Brillouin zone. Isoelectronic substitution breaks some spatial symmetries, causing the Dirac cones to become anisotropic and unpinned from the K point. It also leads to the opening of a small energy gap between the Dirac cone states. In contrast, the nature of the NFE-like parabolic dispersion remains nearly unaltered by the crystal symmetry. Notably, the minimum energy of the NFE band and the effective mass of the electron can be tuned by the composition of the ternary monolayers. Detailed orbital analysis reveals that the contributions of s and pz orbitals of C atoms play a crucial role in tuning the NFE states. Overall, we reveal the novel and versatile electronic properties of the ternary monolayers and demonstrate their tunability via composition.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
