Alyamama Kousa, Reem Ahmed, Mohammad Baraa Abu Bakr, Alaa Nouri Aldosh, Basheer Khalil
{"title":"Complex MEFV and MVK Variations in a Syrian Child: Implications for Clinical Phenotypes and Treatment Response-A Case Report.","authors":"Alyamama Kousa, Reem Ahmed, Mohammad Baraa Abu Bakr, Alaa Nouri Aldosh, Basheer Khalil","doi":"10.1177/23247096241291929","DOIUrl":null,"url":null,"abstract":"<p><p>This case report presents a 10-year-old Syrian boy with concurrent mutations in the Mediterranean fever (<i>MEFV</i>) and mevalonate kinase (<i>MVK</i>) genes, resulting in overlapping symptoms of Familial Mediterranean Fever (FMF) and Hyperimmunoglobulinemia D syndrome (HIDS), both classified as Periodic Fever Syndromes (PFSs). The co-occurrence of these mutations within a single individual is highly unusual. He presented with pallor, intermittent fever, and recurrent respiratory infections from an early age, along with anemia, splenomegaly, hepatomegaly, cervical lymphadenopathy, and growth failure noted in initial investigations. Still's disease was initially considered as the most likely differential diagnosis, leading to the initiation of treatment with methylprednisolone; however, the parents did not follow-up with the treatment. The child returned at 5 years old with appendicitis, which was surgically removed, and parents reported recurrent episodes of arthralgia and joint swelling accompanied by nearly daily fever. Although the child experienced delayed motor development, his cognitive abilities were normal. Genetic analysis identified a homozygous likely pathogenic variant in the <i>MVK</i> gene and a heterozygous likely pathogenic variant in the <i>MEFV</i> gene. The child remains reliant on corticosteroids, with limited response to colchicine and improvement noted after transitioning from tocilizumab to infliximab. The latest follow-up demonstrated significant improvement with no fever, joint swelling, or lymphadenopathy; however, signs of growth failure persist. The atypical manifestations observed in this case may indicate a synergistic effect between the 2 mutations, contributing to the overall clinical picture. Therefore, although HIDS may dominate the clinical presentation, we cannot entirely dismiss the possibility that the FMF mutation plays a role in modulating these symptoms.</p>","PeriodicalId":16198,"journal":{"name":"Journal of investigative medicine high impact case reports","volume":"12 ","pages":"23247096241291929"},"PeriodicalIF":0.8000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11528672/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of investigative medicine high impact case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/23247096241291929","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
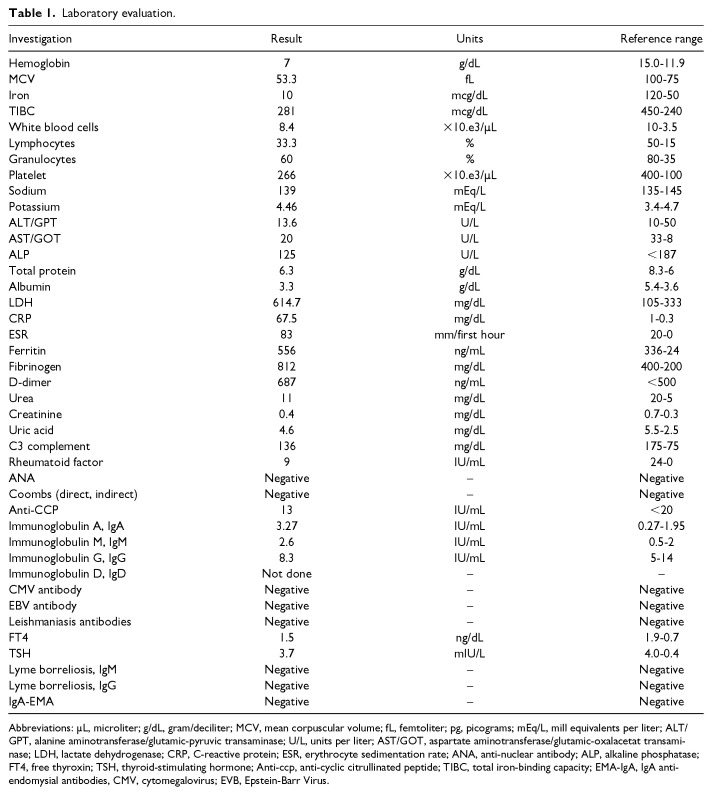
This case report presents a 10-year-old Syrian boy with concurrent mutations in the Mediterranean fever (MEFV) and mevalonate kinase (MVK) genes, resulting in overlapping symptoms of Familial Mediterranean Fever (FMF) and Hyperimmunoglobulinemia D syndrome (HIDS), both classified as Periodic Fever Syndromes (PFSs). The co-occurrence of these mutations within a single individual is highly unusual. He presented with pallor, intermittent fever, and recurrent respiratory infections from an early age, along with anemia, splenomegaly, hepatomegaly, cervical lymphadenopathy, and growth failure noted in initial investigations. Still's disease was initially considered as the most likely differential diagnosis, leading to the initiation of treatment with methylprednisolone; however, the parents did not follow-up with the treatment. The child returned at 5 years old with appendicitis, which was surgically removed, and parents reported recurrent episodes of arthralgia and joint swelling accompanied by nearly daily fever. Although the child experienced delayed motor development, his cognitive abilities were normal. Genetic analysis identified a homozygous likely pathogenic variant in the MVK gene and a heterozygous likely pathogenic variant in the MEFV gene. The child remains reliant on corticosteroids, with limited response to colchicine and improvement noted after transitioning from tocilizumab to infliximab. The latest follow-up demonstrated significant improvement with no fever, joint swelling, or lymphadenopathy; however, signs of growth failure persist. The atypical manifestations observed in this case may indicate a synergistic effect between the 2 mutations, contributing to the overall clinical picture. Therefore, although HIDS may dominate the clinical presentation, we cannot entirely dismiss the possibility that the FMF mutation plays a role in modulating these symptoms.
期刊介绍:
The AFMR is committed to enhancing the training and career development of our members and to furthering its mission to facilitate the conduct of research to improve medical care. Case reports represent an important avenue for trainees (interns, residents, and fellows) and early-stage faculty to demonstrate productive, scholarly activity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
