Exploring 7β-amino-6-nitrocholestens as COVID-19 antivirals: in silico, synthesis, evaluation, and integration of artificial intelligence (AI) in drug design: assessing the cytotoxicity and antioxidant activity of 3β-acetoxynitrocholestane†
Shahabuddin, Uzma, Mohammad Azam, Mehtab Parveen, Nurul Huda Abd Kadir, Kim Min and Mahboob Alam
{"title":"Exploring 7β-amino-6-nitrocholestens as COVID-19 antivirals: in silico, synthesis, evaluation, and integration of artificial intelligence (AI) in drug design: assessing the cytotoxicity and antioxidant activity of 3β-acetoxynitrocholestane†","authors":"Shahabuddin, Uzma, Mohammad Azam, Mehtab Parveen, Nurul Huda Abd Kadir, Kim Min and Mahboob Alam","doi":"10.1039/D4MD00257A","DOIUrl":null,"url":null,"abstract":"<p >In light of the ongoing pandemic caused by SARS-CoV-2, effective and clinically translatable treatments are desperately needed for COVID-19 and its emerging variants. In this study, some derivatives, including 7β-aminocholestene compounds, and 3β-acetoxy-6-nitrocholesta-4,6-diene were synthesized, in quantitative yields from 7β-bromo-6-nitrocholest-5-enes (<strong>1–3</strong>) with a small library of amines. The synthesized steroidal products were then thoroughly characterized using a range of physicochemical techniques, including IR, NMR, UV, MS, and elemental analysis. Next, a virtual screening based on structures using docking studies was conducted to investigate the potential of these synthesized compounds as therapeutic candidates against SARS-CoV-2. Specifically, we evaluated the compounds' binding energy of the reactants and their products with three SARS-CoV-2 functional proteins: the papain-like protease, 3C-like protease or main protease, and RNA-dependent RNA polymerase. Our results indicate that the 7β-aminocholestene derivatives (<strong>4–8</strong>) display intermediate to excellent binding energy, suggesting that they interact strongly with the receptor's active amino acids and may be promising drug candidates for inhibiting SARS-CoV-2. Although the starting steroid derivatives; 7β-bromo-6-nitrocholest-5-enes (<strong>1–3</strong>) and one steroid product; 3β-acetoxy-6-nitrocholesta-4,6-diene (<strong>9</strong>) exhibited strong binding energies with various SARS-CoV-2 receptors, they did not meet the Lipinski Rule and ADMET properties required for drug development. These compounds showed either mutagenic or reproductive/developmental toxicity when assessed using toxicity prediction software. The findings based on structure-based virtual screening, suggest that 7β-aminocholestaines (<strong>4–8</strong>) may be useful for reducing the susceptibility to SARS-CoV-2 infection. The docking pose of compound <strong>4</strong>, which has a high score of −7.4 kcal mol<small><sup>−1</sup></small>, was subjected to AI-assisted deep learning to generate 60 AI-designed molecules for drug design. Molecular docking of these AI molecules was performed to select optimal candidates for further analysis and visualization. The cytotoxicity and antioxidant effects of 3β-acetoxy-6-nitrocholesta-4,6-diene were tested <em>in vitro</em>, showing marked cytotoxicity and antioxidant activity. To elucidate the molecular basis for these effects, steroidal compound 9 was subjected to molecular docking analysis to identify potential binding interactions. The stability of the top-ranked docking pose was subsequently assessed using molecular dynamics simulations.</p>","PeriodicalId":88,"journal":{"name":"MedChemComm","volume":" 11","pages":" 3889-3911"},"PeriodicalIF":3.5970,"publicationDate":"2024-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedChemComm","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/md/d4md00257a","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Pharmacology, Toxicology and Pharmaceutics","Score":null,"Total":0}
引用次数: 0
Abstract
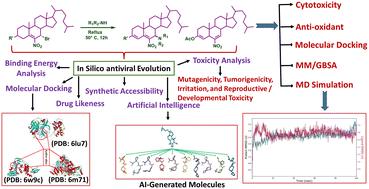
In light of the ongoing pandemic caused by SARS-CoV-2, effective and clinically translatable treatments are desperately needed for COVID-19 and its emerging variants. In this study, some derivatives, including 7β-aminocholestene compounds, and 3β-acetoxy-6-nitrocholesta-4,6-diene were synthesized, in quantitative yields from 7β-bromo-6-nitrocholest-5-enes (1–3) with a small library of amines. The synthesized steroidal products were then thoroughly characterized using a range of physicochemical techniques, including IR, NMR, UV, MS, and elemental analysis. Next, a virtual screening based on structures using docking studies was conducted to investigate the potential of these synthesized compounds as therapeutic candidates against SARS-CoV-2. Specifically, we evaluated the compounds' binding energy of the reactants and their products with three SARS-CoV-2 functional proteins: the papain-like protease, 3C-like protease or main protease, and RNA-dependent RNA polymerase. Our results indicate that the 7β-aminocholestene derivatives (4–8) display intermediate to excellent binding energy, suggesting that they interact strongly with the receptor's active amino acids and may be promising drug candidates for inhibiting SARS-CoV-2. Although the starting steroid derivatives; 7β-bromo-6-nitrocholest-5-enes (1–3) and one steroid product; 3β-acetoxy-6-nitrocholesta-4,6-diene (9) exhibited strong binding energies with various SARS-CoV-2 receptors, they did not meet the Lipinski Rule and ADMET properties required for drug development. These compounds showed either mutagenic or reproductive/developmental toxicity when assessed using toxicity prediction software. The findings based on structure-based virtual screening, suggest that 7β-aminocholestaines (4–8) may be useful for reducing the susceptibility to SARS-CoV-2 infection. The docking pose of compound 4, which has a high score of −7.4 kcal mol−1, was subjected to AI-assisted deep learning to generate 60 AI-designed molecules for drug design. Molecular docking of these AI molecules was performed to select optimal candidates for further analysis and visualization. The cytotoxicity and antioxidant effects of 3β-acetoxy-6-nitrocholesta-4,6-diene were tested in vitro, showing marked cytotoxicity and antioxidant activity. To elucidate the molecular basis for these effects, steroidal compound 9 was subjected to molecular docking analysis to identify potential binding interactions. The stability of the top-ranked docking pose was subsequently assessed using molecular dynamics simulations.
期刊介绍:
Research and review articles in medicinal chemistry and related drug discovery science; the official journal of the European Federation for Medicinal Chemistry.
In 2020, MedChemComm will change its name to RSC Medicinal Chemistry. Issue 12, 2019 will be the last issue as MedChemComm.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
