{"title":"The deficiency of ALKBH5 contributes to hepatic lipid deposition by impairing VPS11-dependent autophagic flux","authors":"Linghuan Li, Yuanhai Sun, Lingqin Li, Wanfang Zheng, Weiwei Zha, Tengjiao Zhao, Guangyao Zhu, Hanbing Li","doi":"10.1111/febs.17299","DOIUrl":null,"url":null,"abstract":"<p>Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease. Hepatic lipid deposition is a key factor in the development of NAFLD. <i>N</i><sup>6</sup>-methyladenosine (m<sup>6</sup>A) modification, the most prevalent mRNA modification in eukaryotic cells, plays an important role in regulating hepatic lipid metabolism. However, its potential role in hepatic lipid deposition remains poorly understood. Histological and immunohistochemistry studies were used to investigate lipid deposition in free fatty acids (FFAs)-incubated LO2 cells, high-fat diet-fed mice models and clinical samples. Stable overexpression and knockdown of AlkB homolog 5 (ALKBH5) was manipulated to investigate the effects of ALKBH5 on m<sup>6</sup>A methylation and lipid metabolism in hepatocytes. RNA-sequencing transcriptome analysis and methylated RNA immunoprecipitation-quantitative-PCR analysis were used to reveal the potential downstream molecular targets of ALKBH5. ALKBH5 was down-regulated in fatty liver compared to normal liver in both humans and mice. Overexpression of ALKBH5 significantly improved FFA-induced lipid accumulation and promoted autophagosome-lysosome fusion in hepatocytes. Meanwhile, knockdown of ALKBH5 significantly increased the expression of microtubule-associated protein 1A/1B-light chain 3B and Sequestosome 1, leading to impaired autophagic flux and further lipid deposition in hepatocytes under FFA incubation. Overexpression of vacuolar protein sorting 11 (VPS11) reversed FFA-induced lipid accumulation in ALKBH5-silenced hepatocytes. Mechanistically, ALKBH5 alleviated hepatic lipid deposition and impaired autophagic flux by removing the m<sup>6</sup>A modification on <i>VPS11</i> mRNA to promote its translation. Collectively, our findings revealed an epigenetic mechanism by which ALKBH5 alleviates hepatic lipid deposition by restoring VPS11-dependent autophagic flux, providing a potential target to counteract NAFLD.</p>","PeriodicalId":94226,"journal":{"name":"The FEBS journal","volume":"291 23","pages":"5256-5275"},"PeriodicalIF":4.2000,"publicationDate":"2024-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The FEBS journal","FirstCategoryId":"1085","ListUrlMain":"https://febs.onlinelibrary.wiley.com/doi/10.1111/febs.17299","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
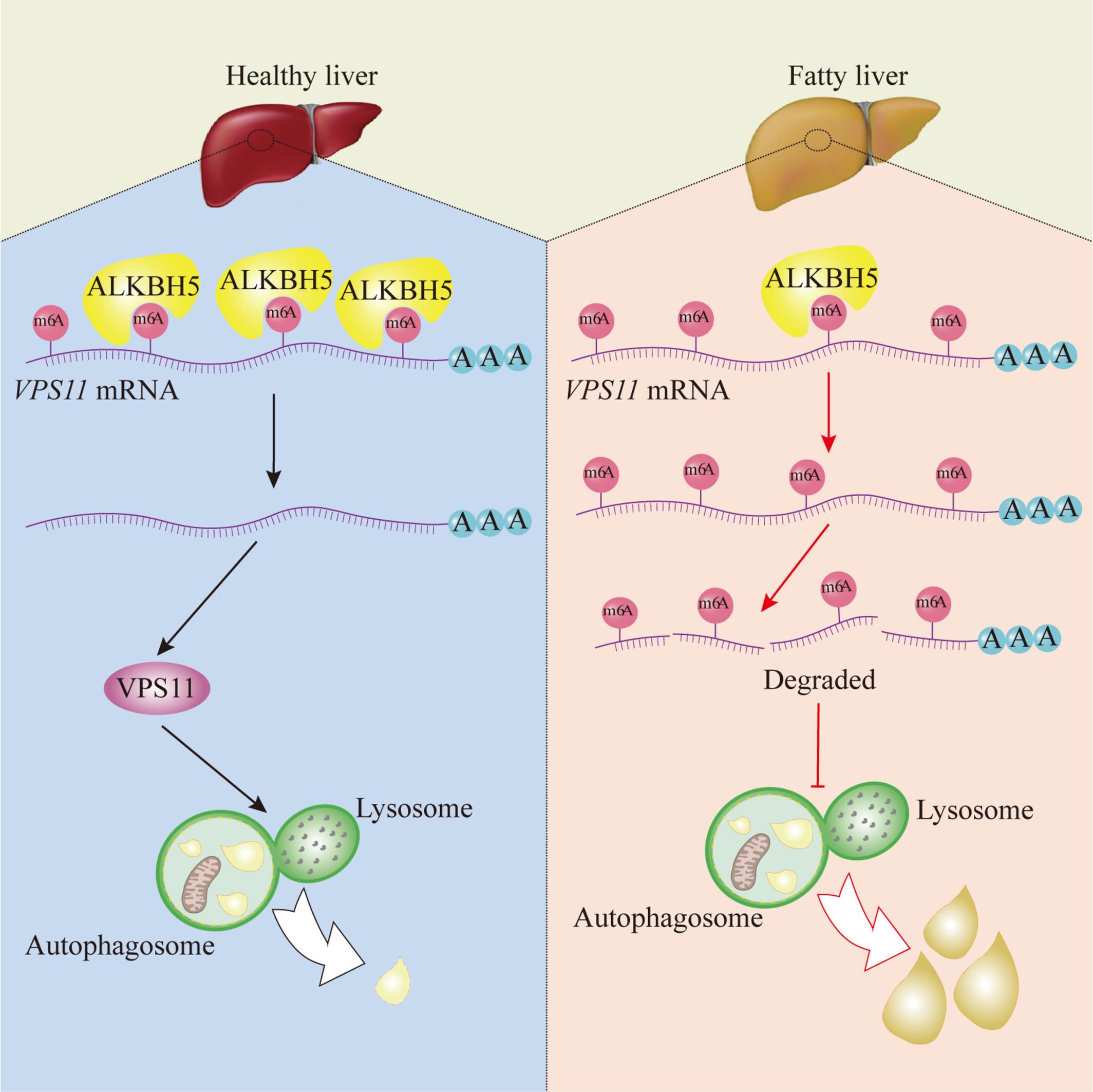
Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease. Hepatic lipid deposition is a key factor in the development of NAFLD. N6-methyladenosine (m6A) modification, the most prevalent mRNA modification in eukaryotic cells, plays an important role in regulating hepatic lipid metabolism. However, its potential role in hepatic lipid deposition remains poorly understood. Histological and immunohistochemistry studies were used to investigate lipid deposition in free fatty acids (FFAs)-incubated LO2 cells, high-fat diet-fed mice models and clinical samples. Stable overexpression and knockdown of AlkB homolog 5 (ALKBH5) was manipulated to investigate the effects of ALKBH5 on m6A methylation and lipid metabolism in hepatocytes. RNA-sequencing transcriptome analysis and methylated RNA immunoprecipitation-quantitative-PCR analysis were used to reveal the potential downstream molecular targets of ALKBH5. ALKBH5 was down-regulated in fatty liver compared to normal liver in both humans and mice. Overexpression of ALKBH5 significantly improved FFA-induced lipid accumulation and promoted autophagosome-lysosome fusion in hepatocytes. Meanwhile, knockdown of ALKBH5 significantly increased the expression of microtubule-associated protein 1A/1B-light chain 3B and Sequestosome 1, leading to impaired autophagic flux and further lipid deposition in hepatocytes under FFA incubation. Overexpression of vacuolar protein sorting 11 (VPS11) reversed FFA-induced lipid accumulation in ALKBH5-silenced hepatocytes. Mechanistically, ALKBH5 alleviated hepatic lipid deposition and impaired autophagic flux by removing the m6A modification on VPS11 mRNA to promote its translation. Collectively, our findings revealed an epigenetic mechanism by which ALKBH5 alleviates hepatic lipid deposition by restoring VPS11-dependent autophagic flux, providing a potential target to counteract NAFLD.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
