Viraj D. Gandhi, Leyan Hua, Morgan Lawrenz, Mohsen Latif, Amber D. Rolland, Iain D. G. Campuzano, Carlos Larriba-Andaluz
{"title":"Elucidating Protein Structures in the Gas Phase: Traversing Configuration Space with Biasing Methods","authors":"Viraj D. Gandhi, Leyan Hua, Morgan Lawrenz, Mohsen Latif, Amber D. Rolland, Iain D. G. Campuzano, Carlos Larriba-Andaluz","doi":"10.1021/acs.jctc.4c00288","DOIUrl":null,"url":null,"abstract":"Achieving accurate characterization of protein structures in the gas phase continues to be a formidable challenge. To tackle this issue, the present study employs Molecular Dynamics (MD) simulations in tandem with enhanced sampling techniques (methods designed to efficiently explore protein conformations). The objective is to identify suitable structures of proteins by contrasting their calculated Collision Cross-Section (CCS) with those observed experimentally. Significant discrepancies were observed between the initial MD-simulated and experimentally measured CCS values through Ion Mobility–Mass Spectrometry (IMS-MS). To bridge this gap, we employed two distinct enhanced sampling methods, Harmonic Biasing Potential and Adaptive Biasing Force, which help the proteins overcome energy barriers to adopt more compact configurations. These techniques leverage the radius of gyration as a reaction coordinate (guiding parameter), guiding the system toward compressed states that potentially match experimental configurations more closely. The guiding forces are only employed to overcome existing barriers and are removed to allow the protein to naturally arrive at a potential gas phase configuration. The results demonstrated close alignment (within ∼4%) between simulated and experimental CCS values despite using different strengths and/or methods, validating their efficacy. This work lays the groundwork for future studies aimed at optimizing biasing methods and expanding the collective variables used for more accurate gas-phase structural predictions.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"9 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00288","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
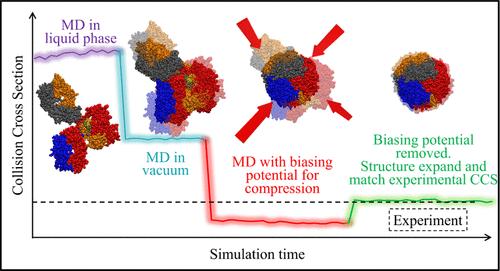
Achieving accurate characterization of protein structures in the gas phase continues to be a formidable challenge. To tackle this issue, the present study employs Molecular Dynamics (MD) simulations in tandem with enhanced sampling techniques (methods designed to efficiently explore protein conformations). The objective is to identify suitable structures of proteins by contrasting their calculated Collision Cross-Section (CCS) with those observed experimentally. Significant discrepancies were observed between the initial MD-simulated and experimentally measured CCS values through Ion Mobility–Mass Spectrometry (IMS-MS). To bridge this gap, we employed two distinct enhanced sampling methods, Harmonic Biasing Potential and Adaptive Biasing Force, which help the proteins overcome energy barriers to adopt more compact configurations. These techniques leverage the radius of gyration as a reaction coordinate (guiding parameter), guiding the system toward compressed states that potentially match experimental configurations more closely. The guiding forces are only employed to overcome existing barriers and are removed to allow the protein to naturally arrive at a potential gas phase configuration. The results demonstrated close alignment (within ∼4%) between simulated and experimental CCS values despite using different strengths and/or methods, validating their efficacy. This work lays the groundwork for future studies aimed at optimizing biasing methods and expanding the collective variables used for more accurate gas-phase structural predictions.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
