Mikuláš Matoušek, Nam Vu, Niranjan Govind, Jonathan J. Foley, IV, Libor Veis
{"title":"Polaritonic Chemistry Using the Density Matrix Renormalization Group Method","authors":"Mikuláš Matoušek, Nam Vu, Niranjan Govind, Jonathan J. Foley, IV, Libor Veis","doi":"10.1021/acs.jctc.4c00986","DOIUrl":null,"url":null,"abstract":"The emerging field of polaritonic chemistry explores the behavior of molecules under strong coupling with cavity modes. Despite recent developments in <i>ab initio</i> polaritonic methods for simulating polaritonic chemistry under electronic strong coupling, their capabilities are limited, especially in cases where the molecule also features strong electronic correlation. To bridge this gap, we have developed a novel method for cavity QED calculations utilizing the Density Matrix Renormalization Group (DMRG) algorithm in conjunction with the Pauli–Fierz Hamiltonian. Our approach is applied to investigate the effect of the cavity on the S<sub>0</sub>–S<sub>1</sub> transition of <i>n</i>-oligoacenes, with <i>n</i> ranging from 2 to 5, encompassing 22 fully correlated π orbitals in the largest pentacene molecule. Our findings indicate that the influence of the cavity intensifies with larger acenes. Additionally, we demonstrate that, unlike the full determinantal representation, DMRG efficiently optimizes and eliminates excess photonic degrees of freedom, resulting in an asymptotically constant computational cost as the photonic basis increases.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"211 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00986","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
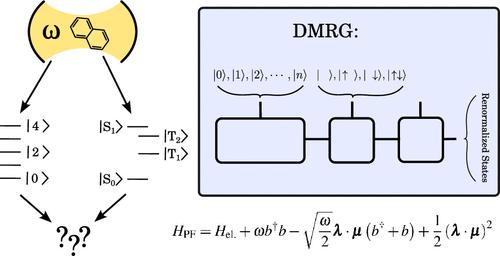
The emerging field of polaritonic chemistry explores the behavior of molecules under strong coupling with cavity modes. Despite recent developments in ab initio polaritonic methods for simulating polaritonic chemistry under electronic strong coupling, their capabilities are limited, especially in cases where the molecule also features strong electronic correlation. To bridge this gap, we have developed a novel method for cavity QED calculations utilizing the Density Matrix Renormalization Group (DMRG) algorithm in conjunction with the Pauli–Fierz Hamiltonian. Our approach is applied to investigate the effect of the cavity on the S0–S1 transition of n-oligoacenes, with n ranging from 2 to 5, encompassing 22 fully correlated π orbitals in the largest pentacene molecule. Our findings indicate that the influence of the cavity intensifies with larger acenes. Additionally, we demonstrate that, unlike the full determinantal representation, DMRG efficiently optimizes and eliminates excess photonic degrees of freedom, resulting in an asymptotically constant computational cost as the photonic basis increases.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
