María Palacios-Ortega, Teresa Guerra-Galán, Adolfo Jiménez-Huete, José María García-Aznar, Marc Pérez-Guzmán, Maria Dolores Mansilla-Ruiz, Ángela Villegas Mendiola, Cristina Pérez López, Elsa Mayol Hornero, Alejandro Peixoto Rodriguez, Ascensión Peña Cortijo, Marta Polo Zarzuela, Marta Mateo Morales, Eduardo Anguita Mandly, Maria Cruz Cárdenas, Alejandra Carrero, Carlos Jiménez García, Estefanía Bolaños, Belén Íñigo, Fiorella Medina, Eduardo de la Fuente, Juliana Ochoa-Grullón, Blanca García-Solís, Yolanda García-Carmona, Miguel Fernández-Arquero, Celina Benavente-Cuesta, Rebeca Pérez de Diego, Nicholas Rider, Silvia Sánchez-Ramón
{"title":"Dissecting Secondary Immunodeficiency: Identification of Primary Immunodeficiency within B-Cell Lymphoproliferative Disorders.","authors":"María Palacios-Ortega, Teresa Guerra-Galán, Adolfo Jiménez-Huete, José María García-Aznar, Marc Pérez-Guzmán, Maria Dolores Mansilla-Ruiz, Ángela Villegas Mendiola, Cristina Pérez López, Elsa Mayol Hornero, Alejandro Peixoto Rodriguez, Ascensión Peña Cortijo, Marta Polo Zarzuela, Marta Mateo Morales, Eduardo Anguita Mandly, Maria Cruz Cárdenas, Alejandra Carrero, Carlos Jiménez García, Estefanía Bolaños, Belén Íñigo, Fiorella Medina, Eduardo de la Fuente, Juliana Ochoa-Grullón, Blanca García-Solís, Yolanda García-Carmona, Miguel Fernández-Arquero, Celina Benavente-Cuesta, Rebeca Pérez de Diego, Nicholas Rider, Silvia Sánchez-Ramón","doi":"10.1007/s10875-024-01818-2","DOIUrl":null,"url":null,"abstract":"<p><p>Distinguishing between primary (PID) and secondary (SID) immunodeficiencies, particularly in relation to hematological B-cell lymphoproliferative disorders (B-CLPD), poses a major clinical challenge. We aimed to analyze and define the clinical and laboratory variables in SID patients associated with B-CLPD, identifying overlaps with late-onset PIDs, which could potentially improve diagnostic precision and prognostic assessment. We studied 37 clinical/laboratory variables in 151 SID patients with B-CLPD. Patients were classified as \"Suspected PID Group\" when having recurrent-severe infections prior to the B-CLPD and/or hypogammaglobulinemia according to key ESID criteria for PID. Bivariate association analyses showed significant statistical differences between \"Suspected PID\"- and \"SID\"-groups in 10 out of 37 variables analyzed, with \"Suspected PID\" showing higher frequencies of childhood recurrent-severe infections, family history of B-CLPD, significantly lower serum Free Light Chain (sFLC), immunoglobulin concentrations, lower total leukocyte, and switch-memory B-cell counts at baseline. Rpart machine learning algorithm was performed to potentially create a model to differentiate both groups. The model developed a decision tree with two major variables in order of relevance: sum κ + λ and history of severe-recurrent infections in childhood, with high sensitivity 89.5%, specificity 100%, and accuracy 91.8% for PID prediction. Identifying significant clinical and immunological variables can aid in the difficult task of recognizing late-onset PIDs among SID patients, emphasizing the value of a comprehensive immunological evaluation. The differences between \"Suspected PID\" and SID groups, highlight the need of early, tailored diagnostic and treatment strategies for personalized patient management and follow up.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"32"},"PeriodicalIF":5.7000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11499357/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-024-01818-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
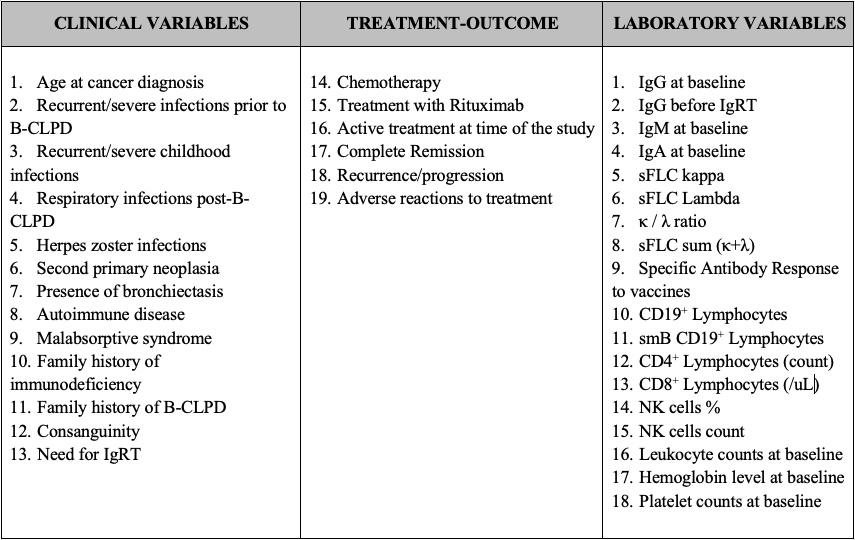
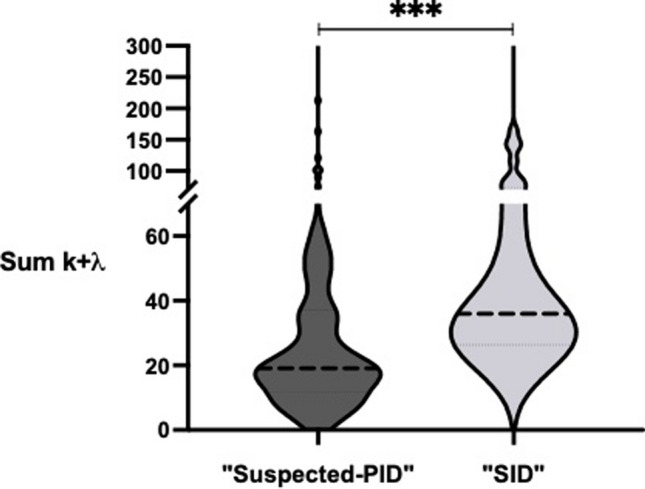
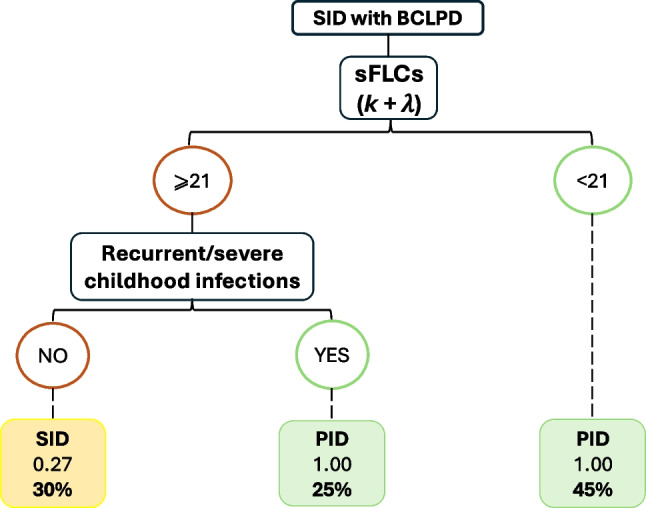
Distinguishing between primary (PID) and secondary (SID) immunodeficiencies, particularly in relation to hematological B-cell lymphoproliferative disorders (B-CLPD), poses a major clinical challenge. We aimed to analyze and define the clinical and laboratory variables in SID patients associated with B-CLPD, identifying overlaps with late-onset PIDs, which could potentially improve diagnostic precision and prognostic assessment. We studied 37 clinical/laboratory variables in 151 SID patients with B-CLPD. Patients were classified as "Suspected PID Group" when having recurrent-severe infections prior to the B-CLPD and/or hypogammaglobulinemia according to key ESID criteria for PID. Bivariate association analyses showed significant statistical differences between "Suspected PID"- and "SID"-groups in 10 out of 37 variables analyzed, with "Suspected PID" showing higher frequencies of childhood recurrent-severe infections, family history of B-CLPD, significantly lower serum Free Light Chain (sFLC), immunoglobulin concentrations, lower total leukocyte, and switch-memory B-cell counts at baseline. Rpart machine learning algorithm was performed to potentially create a model to differentiate both groups. The model developed a decision tree with two major variables in order of relevance: sum κ + λ and history of severe-recurrent infections in childhood, with high sensitivity 89.5%, specificity 100%, and accuracy 91.8% for PID prediction. Identifying significant clinical and immunological variables can aid in the difficult task of recognizing late-onset PIDs among SID patients, emphasizing the value of a comprehensive immunological evaluation. The differences between "Suspected PID" and SID groups, highlight the need of early, tailored diagnostic and treatment strategies for personalized patient management and follow up.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
