{"title":"m6A demethylase FTO transcriptionally activated by SP1 improves ischemia reperfusion-triggered acute kidney injury by activating Ambra1/ULK1-mediated autophagy","authors":"Yan Chen, Yuanfei Liu, Weiping Tu, Yanxia Chen, Chengyun Xu, Chong Huang","doi":"10.1096/fj.202400132RRR","DOIUrl":null,"url":null,"abstract":"<p>Ischemia reperfusion (I/R) was considered as one of main causes of acute kidney injury (AKI). However, the exact mechanism remains unclear. Here, this study aimed to investigate the role and mechanism of the m6A demethylase fat mass and obesity-associated (FTO) protein in I/R-induced AKI. HK-2 cells and SD rats were utilized to establish hypoxia/reoxygenation (H/R) or I/R induced AKI models. The changes of RNAs and proteins were quantified using RT-qPCR, western blot, and immunofluorescence assays, respectively. Cell proliferation and apoptosis were assessed by CCK-8 and flow cytometry. Interactions between molecules were investigated using RIP, ChIP, Co-IP, RNA pull-down, and dual luciferase reporter assays. Global m6A quantification was evaluated by kits. TUNEL and HE staining were employed for histopathological examinations. Oxidative stress-related indicators and renal function were determined using ELISA assays. The FTO expression was downregulated in H/R-induced HK-2 cells and renal tissues from I/R-induced rats. Overexpression of FTO improved the cell viability but repressed apoptosis and oxidative stress in H/R-treated HK-2 cells, as well as enhanced renal function and alleviated kidney injury in I/R rats. Notably, the FTO overexpression significantly increased autophagy-related LC3 and ULK1 levels. When autophagy was inhibited, the protective effects of FTO in AKI were diminished. Notably, Ambra1, a crucial regulator of autophagy, was repressed in H/R-induced HK-2 cells. However, the FTO overexpression restored the Ambra1 expression by reducing m6A modification of its mRNA. SP1, acting as an upstream transcription factor, directly interacts with the FTO promoter to enhance FTO expression. Knockdown of SP1 or Ambra1 suppressed the beneficial effects of FTO upregulation on autophagy and oxidative stress injury in H/R-stimulated cells. FTO, transcriptionally activated by SP1, promoted autophagy by upregulating Ambra1/ULK1 signaling, thereby inhibiting oxidative stress and kidney injury. These findings may provide some novel insights for AKI treatment.</p>","PeriodicalId":50455,"journal":{"name":"The FASEB Journal","volume":"38 20","pages":""},"PeriodicalIF":4.2000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1096/fj.202400132RRR","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The FASEB Journal","FirstCategoryId":"99","ListUrlMain":"https://faseb.onlinelibrary.wiley.com/doi/10.1096/fj.202400132RRR","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
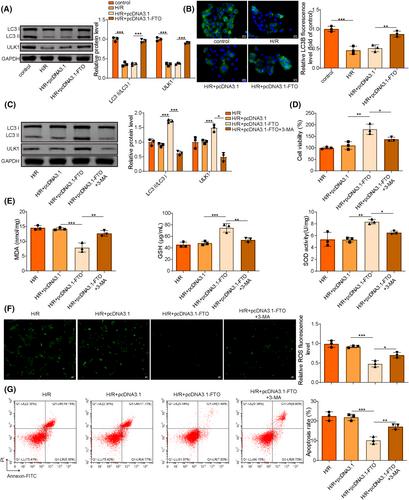
Ischemia reperfusion (I/R) was considered as one of main causes of acute kidney injury (AKI). However, the exact mechanism remains unclear. Here, this study aimed to investigate the role and mechanism of the m6A demethylase fat mass and obesity-associated (FTO) protein in I/R-induced AKI. HK-2 cells and SD rats were utilized to establish hypoxia/reoxygenation (H/R) or I/R induced AKI models. The changes of RNAs and proteins were quantified using RT-qPCR, western blot, and immunofluorescence assays, respectively. Cell proliferation and apoptosis were assessed by CCK-8 and flow cytometry. Interactions between molecules were investigated using RIP, ChIP, Co-IP, RNA pull-down, and dual luciferase reporter assays. Global m6A quantification was evaluated by kits. TUNEL and HE staining were employed for histopathological examinations. Oxidative stress-related indicators and renal function were determined using ELISA assays. The FTO expression was downregulated in H/R-induced HK-2 cells and renal tissues from I/R-induced rats. Overexpression of FTO improved the cell viability but repressed apoptosis and oxidative stress in H/R-treated HK-2 cells, as well as enhanced renal function and alleviated kidney injury in I/R rats. Notably, the FTO overexpression significantly increased autophagy-related LC3 and ULK1 levels. When autophagy was inhibited, the protective effects of FTO in AKI were diminished. Notably, Ambra1, a crucial regulator of autophagy, was repressed in H/R-induced HK-2 cells. However, the FTO overexpression restored the Ambra1 expression by reducing m6A modification of its mRNA. SP1, acting as an upstream transcription factor, directly interacts with the FTO promoter to enhance FTO expression. Knockdown of SP1 or Ambra1 suppressed the beneficial effects of FTO upregulation on autophagy and oxidative stress injury in H/R-stimulated cells. FTO, transcriptionally activated by SP1, promoted autophagy by upregulating Ambra1/ULK1 signaling, thereby inhibiting oxidative stress and kidney injury. These findings may provide some novel insights for AKI treatment.
期刊介绍:
The FASEB Journal publishes international, transdisciplinary research covering all fields of biology at every level of organization: atomic, molecular, cell, tissue, organ, organismic and population. While the journal strives to include research that cuts across the biological sciences, it also considers submissions that lie within one field, but may have implications for other fields as well. The journal seeks to publish basic and translational research, but also welcomes reports of pre-clinical and early clinical research. In addition to research, review, and hypothesis submissions, The FASEB Journal also seeks perspectives, commentaries, book reviews, and similar content related to the life sciences in its Up Front section.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
