Pengfei Ma , Rui Wang , Chengsong Liu , Pengfei Liu , Chaozheng He , Wei Song , Tao Zhang
{"title":"Theoretical insights into efficient electrocatalysts for nitrogen reduction reaction by transition metal atoms supported on g-C3N4","authors":"Pengfei Ma , Rui Wang , Chengsong Liu , Pengfei Liu , Chaozheng He , Wei Song , Tao Zhang","doi":"10.1016/j.comptc.2024.114932","DOIUrl":null,"url":null,"abstract":"<div><div>NH<sub>3</sub> is considered one of the most important chemicals, not only as an industrial feedstock for the production of fertilizers, but also as renewable carbon-free energy carrier. In recent years, electrocatalytic nitrogen reduction reaction (NRR), which uses N<sub>2</sub> and H<sub>2</sub>O as raw materials to synthesize NH<sub>3</sub>, is considered as one of the most promising methods for N<sub>2</sub> fixation. Therefore, the design and synthesis of NRR electrocatalysts with high catalytic performance is very important. Herein, using density functional theory method, 3d transition metal single atom anchored to g-C<sub>3</sub>N<sub>4</sub> as single-atom catalysts was studied for the catalytic reduction of N<sub>2</sub>. The results found that V@g-C<sub>3</sub>N<sub>4</sub> exhibited high electrocatalytic activity and good catalytic selectivity. The limiting potential was only –0.115 V. Furthermore, the relevant electronic properties of V@g-C<sub>3</sub>N<sub>4</sub> were also analyzed. Our work can provide ideas and help to understand the reaction mechanism of single atom doped g-C<sub>3</sub>N<sub>4</sub> as NRR electrocatalysts with excellent catalytic performance.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114932"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004717","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/22 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
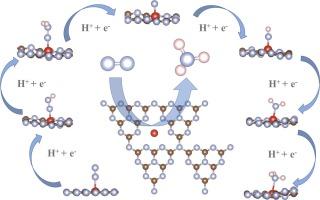
NH3 is considered one of the most important chemicals, not only as an industrial feedstock for the production of fertilizers, but also as renewable carbon-free energy carrier. In recent years, electrocatalytic nitrogen reduction reaction (NRR), which uses N2 and H2O as raw materials to synthesize NH3, is considered as one of the most promising methods for N2 fixation. Therefore, the design and synthesis of NRR electrocatalysts with high catalytic performance is very important. Herein, using density functional theory method, 3d transition metal single atom anchored to g-C3N4 as single-atom catalysts was studied for the catalytic reduction of N2. The results found that V@g-C3N4 exhibited high electrocatalytic activity and good catalytic selectivity. The limiting potential was only –0.115 V. Furthermore, the relevant electronic properties of V@g-C3N4 were also analyzed. Our work can provide ideas and help to understand the reaction mechanism of single atom doped g-C3N4 as NRR electrocatalysts with excellent catalytic performance.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
