{"title":"Integrated all-atom and coarse-grained simulations uncover structural, dynamics and energetic shifts in SARS-CoV-2 JN.1 and BA.2.86 variants","authors":"Akshit Sharma , Shweata Maurya , Timir Tripathi , Aditya K. Padhi","doi":"10.1016/j.actatropica.2024.107444","DOIUrl":null,"url":null,"abstract":"<div><div>Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), responsible for the COVID-19 pandemic, is an enveloped, positive-stranded RNA virus that enters human cells by using its spike protein to bind to the human angiotensin-converting enzyme 2 (ACE2) receptor. Since its emergence, the virus has mutated, producing variants with increased transmissibility, immune evasion, and infectivity. The JN.1 variant, detected in January 2024, features a single substitution mutation (Leu455Ser) in the receptor-binding domain (RBD) of its spike protein, setting it apart from its parent lineage, BA.2.86. This variant has rapidly become globally predominant due to its enhanced transmission and significant epidemiological impact. To understand the causes behind the dominance of the JN.1 variant, we conducted a comprehensive study using all-atom molecular dynamics (MD) and coarse-grained MD simulations. This allowed us to examine the structural, dynamic, energetics and binding properties of the wild-type (Wuhan strain), BA.2.86, and JN.1 variants. Principal component and free energy landscape analyses revealed enhanced structural stability in the JN.1 variant. Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) assessments indicated lower binding affinity for JN.1 as compared to BA.2.86. Intermolecular interaction analyses further confirmed BA.2.86′s superior binding affinity over JN.1 and wild-type. Additionally, we compared and validated our findings against experimentally determined cryo-electron microscopy (cryo-EM) structures of JN.1 and BA.2.86 variants, confirming the reliability of our simulation results. Overall, this study provides crucial insights into the structural-dynamics-energetics features and physicochemical properties that have contributed to the global prevalence of the JN.1 variant and sheds light on its potential to generate future subvariants.</div></div>","PeriodicalId":7240,"journal":{"name":"Acta tropica","volume":"260 ","pages":"Article 107444"},"PeriodicalIF":2.6000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta tropica","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0001706X24003255","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/28 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"PARASITOLOGY","Score":null,"Total":0}
引用次数: 0
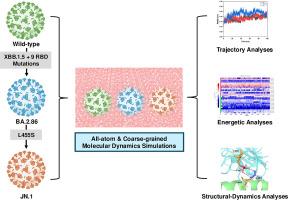
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), responsible for the COVID-19 pandemic, is an enveloped, positive-stranded RNA virus that enters human cells by using its spike protein to bind to the human angiotensin-converting enzyme 2 (ACE2) receptor. Since its emergence, the virus has mutated, producing variants with increased transmissibility, immune evasion, and infectivity. The JN.1 variant, detected in January 2024, features a single substitution mutation (Leu455Ser) in the receptor-binding domain (RBD) of its spike protein, setting it apart from its parent lineage, BA.2.86. This variant has rapidly become globally predominant due to its enhanced transmission and significant epidemiological impact. To understand the causes behind the dominance of the JN.1 variant, we conducted a comprehensive study using all-atom molecular dynamics (MD) and coarse-grained MD simulations. This allowed us to examine the structural, dynamic, energetics and binding properties of the wild-type (Wuhan strain), BA.2.86, and JN.1 variants. Principal component and free energy landscape analyses revealed enhanced structural stability in the JN.1 variant. Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) assessments indicated lower binding affinity for JN.1 as compared to BA.2.86. Intermolecular interaction analyses further confirmed BA.2.86′s superior binding affinity over JN.1 and wild-type. Additionally, we compared and validated our findings against experimentally determined cryo-electron microscopy (cryo-EM) structures of JN.1 and BA.2.86 variants, confirming the reliability of our simulation results. Overall, this study provides crucial insights into the structural-dynamics-energetics features and physicochemical properties that have contributed to the global prevalence of the JN.1 variant and sheds light on its potential to generate future subvariants.
期刊介绍:
Acta Tropica, is an international journal on infectious diseases that covers public health sciences and biomedical research with particular emphasis on topics relevant to human and animal health in the tropics and the subtropics.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
