Charalampos G. Livas, Pantelis N. Trikalitis, George E. Froudakis
{"title":"MOFSynth: A Computational Tool toward Synthetic Likelihood Predictions of MOFs","authors":"Charalampos G. Livas, Pantelis N. Trikalitis, George E. Froudakis","doi":"10.1021/acs.jcim.4c01298","DOIUrl":null,"url":null,"abstract":"In the past decade, high-throughput computational studies of materials have increased significantly mainly due to advances in computer capabilities and have attracted a great deal of interest. In the field of metal–organic frameworks (MOFs), over a million hypothetical MOFs have been designed in silico, yet only a small fraction of these have been synthesized. For validating the computational-hypothetical results and accelerating the progress in the field, there is a pressing need for distinguishing MOFs that are more likely to be synthesized for real-life applications. This study presents a comprehensive investigation into the synthesizability likelihood of MOFs, utilizing a novel computational approach based on the disparities in energy and geometry between the linker conformation within the MOF structure and its isolated, free-gas state since both of these have been proven to be critical factors influencing MOF synthesis. Our user-friendly tool streamlines synthesizability evaluation, requiring minimal expertise in computational chemistry. By deconstructing over 40,000 MOFs from databases, including QMOF, CoRE MOF, and ToBaCCo, we analyze key parameters defining the linker strain within the MOF unit cell. Our results indicate that QMOF and CoRE MOF contain more promising candidates for synthesis, while ToBaCCo exhibits a relatively poor synthesizability likelihood due to unoptimized materials. Through extensive analysis, we identify optimal linker candidates for highly synthesizable MOFs. Consistent trends in energy distribution across databases that are confirmed by high Pearson and Spearman coefficients suggest the potential for omitting optimization calculations, significantly reducing computational costs. This study underscores the importance of linker deformation and energy disparities and enhances our understanding of synthetic accessibility in MOF research, offering valuable insights for future advancements in the field.","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"43 1","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2024-10-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c01298","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
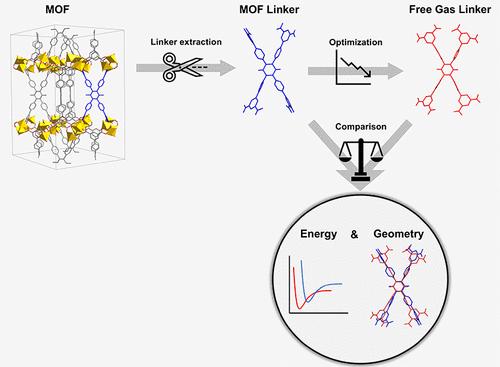
In the past decade, high-throughput computational studies of materials have increased significantly mainly due to advances in computer capabilities and have attracted a great deal of interest. In the field of metal–organic frameworks (MOFs), over a million hypothetical MOFs have been designed in silico, yet only a small fraction of these have been synthesized. For validating the computational-hypothetical results and accelerating the progress in the field, there is a pressing need for distinguishing MOFs that are more likely to be synthesized for real-life applications. This study presents a comprehensive investigation into the synthesizability likelihood of MOFs, utilizing a novel computational approach based on the disparities in energy and geometry between the linker conformation within the MOF structure and its isolated, free-gas state since both of these have been proven to be critical factors influencing MOF synthesis. Our user-friendly tool streamlines synthesizability evaluation, requiring minimal expertise in computational chemistry. By deconstructing over 40,000 MOFs from databases, including QMOF, CoRE MOF, and ToBaCCo, we analyze key parameters defining the linker strain within the MOF unit cell. Our results indicate that QMOF and CoRE MOF contain more promising candidates for synthesis, while ToBaCCo exhibits a relatively poor synthesizability likelihood due to unoptimized materials. Through extensive analysis, we identify optimal linker candidates for highly synthesizable MOFs. Consistent trends in energy distribution across databases that are confirmed by high Pearson and Spearman coefficients suggest the potential for omitting optimization calculations, significantly reducing computational costs. This study underscores the importance of linker deformation and energy disparities and enhances our understanding of synthetic accessibility in MOF research, offering valuable insights for future advancements in the field.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
