Peter Škorňa , Sanam Bashir , Eva Scholtzová , Daniel Tunega
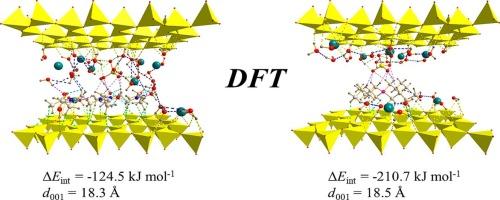
{"title":"Model study on potential removal of toxic Se(VI) by organically modified montmorillonite","authors":"Peter Škorňa , Sanam Bashir , Eva Scholtzová , Daniel Tunega","doi":"10.1016/j.comptc.2024.114939","DOIUrl":null,"url":null,"abstract":"<div><div>The theoretical DFT-D3 approach was used in the model study of the immobilisation of the toxic selenate oxyanion, with montmorillonite clay modified by poly(2-methyl-2-oxazoline) polymer represented by a pentamer unit, and tetrabutylphosphonium cation. The calculated basal spacing for Se-POx-Mt and Se-TBP-Mt models provided similar results differing by 0.2 Å (18.3 Å for Se-POx-Mt and/or 18.5 Å for Se-TBP-Mt, respectively). The calculated intercalation energy, Δ<em>E</em><sub>int,</sub> showed a suitability of both modified Mts for trapping of (SeO<sub>4</sub>)<sup>2–</sup> oxyanions favouring the Se-TBP-Mt (–210.7 kJ/mol) system compared to Se-POx-Mt (–124.5 kJ/mol). The main interactions in both models were classified as hydrogen bonds of weak (C<img>H···O) and moderate-to-strong (O<img>H···O) strength. The calculated total and projected vibrational density of states of the Se-POx-Mt and Se‑TBP‑Mt models, obtained from the <em>ab initio</em> molecular dynamics simulations, were used for distinct identification of vibrational modes of the intercalated (SeO<sub>4</sub>)<sup>2–</sup> ion in the interlayer space of both models.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114939"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X2400478X","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/24 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
The theoretical DFT-D3 approach was used in the model study of the immobilisation of the toxic selenate oxyanion, with montmorillonite clay modified by poly(2-methyl-2-oxazoline) polymer represented by a pentamer unit, and tetrabutylphosphonium cation. The calculated basal spacing for Se-POx-Mt and Se-TBP-Mt models provided similar results differing by 0.2 Å (18.3 Å for Se-POx-Mt and/or 18.5 Å for Se-TBP-Mt, respectively). The calculated intercalation energy, ΔEint, showed a suitability of both modified Mts for trapping of (SeO4)2– oxyanions favouring the Se-TBP-Mt (–210.7 kJ/mol) system compared to Se-POx-Mt (–124.5 kJ/mol). The main interactions in both models were classified as hydrogen bonds of weak (CH···O) and moderate-to-strong (OH···O) strength. The calculated total and projected vibrational density of states of the Se-POx-Mt and Se‑TBP‑Mt models, obtained from the ab initio molecular dynamics simulations, were used for distinct identification of vibrational modes of the intercalated (SeO4)2– ion in the interlayer space of both models.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
