Negar Safinianaini , Camila P.E. De Souza , Andrew Roth , Hazal Koptagel , Hosein Toosi , Jens Lagergren
{"title":"CopyMix: Mixture model based single-cell clustering and copy number profiling using variational inference","authors":"Negar Safinianaini , Camila P.E. De Souza , Andrew Roth , Hazal Koptagel , Hosein Toosi , Jens Lagergren","doi":"10.1016/j.compbiolchem.2024.108257","DOIUrl":null,"url":null,"abstract":"<div><div>Investigating tumor heterogeneity using single-cell sequencing technologies is imperative to understand how tumors evolve since each cell subpopulation harbors a unique set of genomic features that yields a unique phenotype, which is bound to have clinical relevance. Clustering of cells based on copy number data obtained from single-cell DNA sequencing provides an opportunity to identify different tumor cell subpopulations. Accordingly, computational methods have emerged for single-cell copy number profiling and clustering; however, these two tasks have been handled sequentially by applying various ad-hoc pre- and post-processing steps; hence, a procedure vulnerable to introducing clustering artifacts. We avoid the clustering artifact issues in our method, CopyMix, a Variational Inference for a novel mixture model, by jointly inferring cell clusters and their underlying copy number profile. Our probabilistic graphical model is an improved version of the mixture of hidden Markov models, which is designed uniquely to infer single-cell copy number profiling and clustering. For the evaluation, we used likelihood-ratio test, CH index, Silhouette, V-measure, total variation scores. CopyMix performs well on both biological and simulated data. Our favorable results indicate a considerable potential to obtain clinical impact by using CopyMix in studies of cancer tumor heterogeneity.</div></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":"113 ","pages":"Article 108257"},"PeriodicalIF":3.1000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124002457","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/23 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
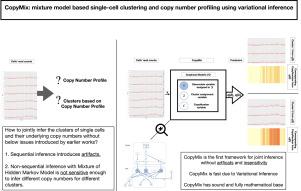
Investigating tumor heterogeneity using single-cell sequencing technologies is imperative to understand how tumors evolve since each cell subpopulation harbors a unique set of genomic features that yields a unique phenotype, which is bound to have clinical relevance. Clustering of cells based on copy number data obtained from single-cell DNA sequencing provides an opportunity to identify different tumor cell subpopulations. Accordingly, computational methods have emerged for single-cell copy number profiling and clustering; however, these two tasks have been handled sequentially by applying various ad-hoc pre- and post-processing steps; hence, a procedure vulnerable to introducing clustering artifacts. We avoid the clustering artifact issues in our method, CopyMix, a Variational Inference for a novel mixture model, by jointly inferring cell clusters and their underlying copy number profile. Our probabilistic graphical model is an improved version of the mixture of hidden Markov models, which is designed uniquely to infer single-cell copy number profiling and clustering. For the evaluation, we used likelihood-ratio test, CH index, Silhouette, V-measure, total variation scores. CopyMix performs well on both biological and simulated data. Our favorable results indicate a considerable potential to obtain clinical impact by using CopyMix in studies of cancer tumor heterogeneity.
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
