Annabelle L Willemsen, David J Torpy, Sunita M C De Sousa, Henrik Falhammar, R Louise Rushworth
{"title":"17α-Hydroxylase/17,20-lyase Deficiency (17-OHD): A Meta-analysis of Reported Cases.","authors":"Annabelle L Willemsen, David J Torpy, Sunita M C De Sousa, Henrik Falhammar, R Louise Rushworth","doi":"10.1210/clinem/dgae773","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>Homozygous pathogenic variants in the CYP17A1 gene result in defective activity of the steroidogenic enzymes 17α-hydroxylase/17,20-lyase resulting in the clinical syndrome 17-OHD characterized by hypertension, hypokalemia, and disorders of sexual development. Pathogenic variants of CYP17A1 lead to complete or partial loss of enzymatic activity and clinical presentations of varying severity. This study aimed to examine relationships between CYP17A1 genotype and clinical presentation in a global cohort.</p><p><strong>Methods: </strong>We searched PubMed and Scopus for case reports and cohort studies reporting clinical data on patients with 17-OHD published between 1988 and 2022. Of 451 studies, 178 met inclusion criteria comprising a total of 465 patients. We pooled patient data and examined associations between causative variants and their clinical presentations.</p><p><strong>Results: </strong>There were 465 unique patients with a mean age of 18.9 (9.0) years, 52.5% (n = 244) were XY and 6.4% (n = 29) were phenotypically male. Homozygous variants were seen in 48.0% (n = 223) of patients. Common clinical presentations were hypertension (57.0%, n = 256), hypokalemia (45.4% n = 211), primary amenorrhea (38.3%, n = 178), cryptorchidism (15.3%, n = 71), and atypical genitalia (14.2%, n = 66). Frequently occurring variants included p.Y329Kfs (n = 86), p.D487_F489del (n = 44), and p.W406R (n = 39). More severe variants, such as p.Y329Kfs, were associated with hypocortisolism (P < .05), combined hypokalemia and hypertension (P < .01), and disordered sexual development (P < .01).</p><p><strong>Main conclusion: </strong>17-OHD is a rare, frequently misdiagnosed disease. Male patients are typically diagnosed earlier because of genital dysplasia associated with less severe variants, whereas female patients are typically diagnosed later from primary amenorrhea and hypertension. Patients presenting with disordered sexual development and hypertension should be investigated for 17-OHD.</p>","PeriodicalId":50238,"journal":{"name":"Journal of Clinical Endocrinology & Metabolism","volume":" ","pages":"e1261-e1271"},"PeriodicalIF":5.1000,"publicationDate":"2025-03-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11913080/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Endocrinology & Metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1210/clinem/dgae773","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
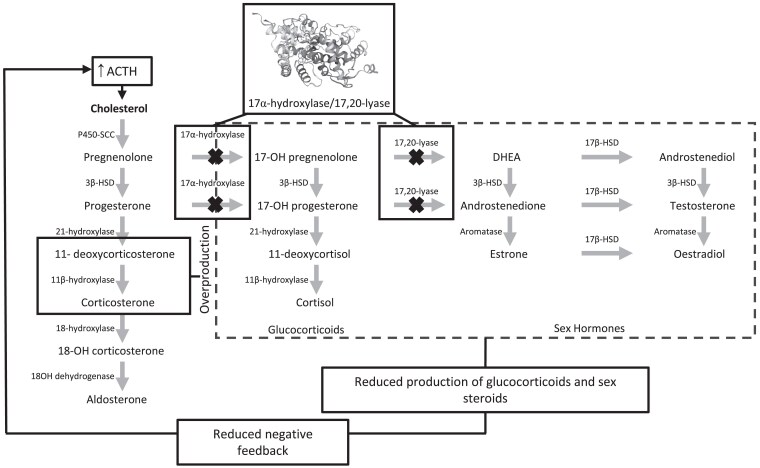
Purpose: Homozygous pathogenic variants in the CYP17A1 gene result in defective activity of the steroidogenic enzymes 17α-hydroxylase/17,20-lyase resulting in the clinical syndrome 17-OHD characterized by hypertension, hypokalemia, and disorders of sexual development. Pathogenic variants of CYP17A1 lead to complete or partial loss of enzymatic activity and clinical presentations of varying severity. This study aimed to examine relationships between CYP17A1 genotype and clinical presentation in a global cohort.
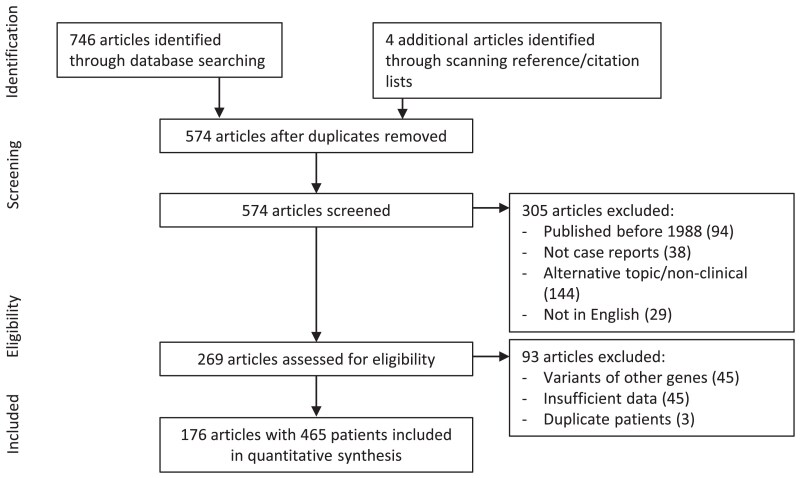
Methods: We searched PubMed and Scopus for case reports and cohort studies reporting clinical data on patients with 17-OHD published between 1988 and 2022. Of 451 studies, 178 met inclusion criteria comprising a total of 465 patients. We pooled patient data and examined associations between causative variants and their clinical presentations.
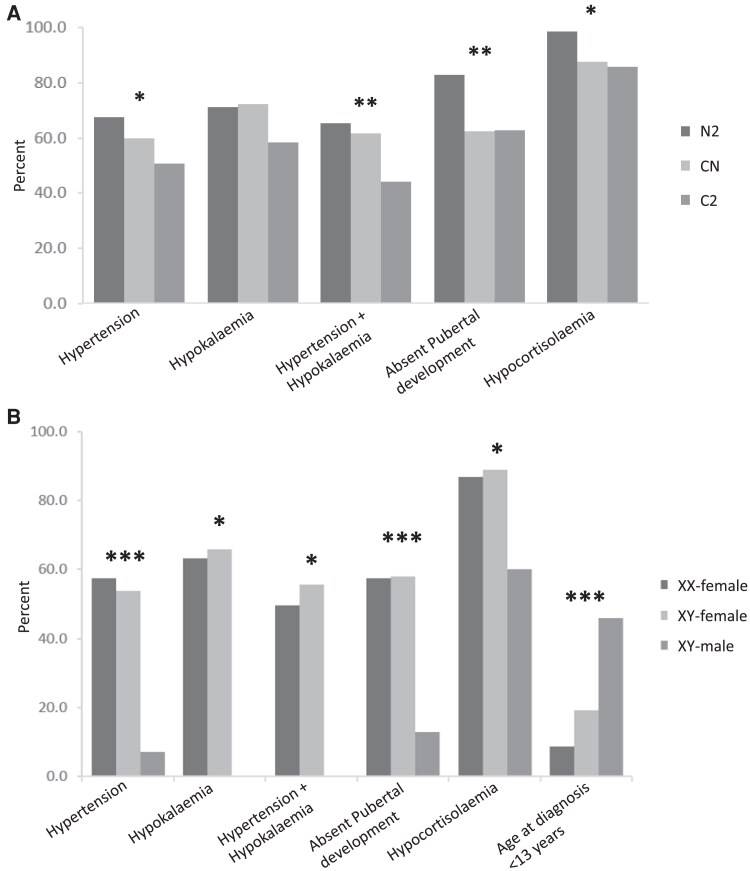
Results: There were 465 unique patients with a mean age of 18.9 (9.0) years, 52.5% (n = 244) were XY and 6.4% (n = 29) were phenotypically male. Homozygous variants were seen in 48.0% (n = 223) of patients. Common clinical presentations were hypertension (57.0%, n = 256), hypokalemia (45.4% n = 211), primary amenorrhea (38.3%, n = 178), cryptorchidism (15.3%, n = 71), and atypical genitalia (14.2%, n = 66). Frequently occurring variants included p.Y329Kfs (n = 86), p.D487_F489del (n = 44), and p.W406R (n = 39). More severe variants, such as p.Y329Kfs, were associated with hypocortisolism (P < .05), combined hypokalemia and hypertension (P < .01), and disordered sexual development (P < .01).
Main conclusion: 17-OHD is a rare, frequently misdiagnosed disease. Male patients are typically diagnosed earlier because of genital dysplasia associated with less severe variants, whereas female patients are typically diagnosed later from primary amenorrhea and hypertension. Patients presenting with disordered sexual development and hypertension should be investigated for 17-OHD.
期刊介绍:
The Journal of Clinical Endocrinology & Metabolism is the world"s leading peer-reviewed journal for endocrine clinical research and cutting edge clinical practice reviews. Each issue provides the latest in-depth coverage of new developments enhancing our understanding, diagnosis and treatment of endocrine and metabolic disorders. Regular features of special interest to endocrine consultants include clinical trials, clinical reviews, clinical practice guidelines, case seminars, and controversies in clinical endocrinology, as well as original reports of the most important advances in patient-oriented endocrine and metabolic research. According to the latest Thomson Reuters Journal Citation Report, JCE&M articles were cited 64,185 times in 2008.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
