Masoumeh Mahmoudi Gahrouei, Nikiphoros Vlastos, Oreoluwa Adesina, Laura de Sousa Oliveira
{"title":"Benchmark Investigation of SCC-DFTB Against Standard DFT to Model Phononic Properties in Two-Dimensional MOFs for Thermoelectric Applications.","authors":"Masoumeh Mahmoudi Gahrouei, Nikiphoros Vlastos, Oreoluwa Adesina, Laura de Sousa Oliveira","doi":"10.1021/acs.jctc.4c01229","DOIUrl":null,"url":null,"abstract":"<p><p>Despite the importance of modeling lattice thermal conductivity in predicting thermoelectric (TE) properties, computational data on heat transport, especially from first-principles, in 2D metal-organic frameworks (MOFs) remain limited due to the high computational cost. To address this, we provide a benchmark of the performance of semiempirical self-consistent-charge density functional tight-binding (SCC-DFTB) methods against density functional theory (DFT) for monolayer, serrated, AA-stacked and/or AB-stacked Zn<sub>3</sub>C<sub>6</sub>O<sub>6</sub>, Cd<sub>3</sub>C<sub>6</sub>O<sub>6</sub>, Zn-NH-MOF, and Ni<sub>3</sub>(HITP)<sub>2</sub> MOFs. Harmonic lattice dynamics calculations, including partial atomic contributions to phonon dispersions, are evaluated with both SCC-DFTB and DFT, whereas anharmonic transport (i.e., thermal conductivity) is evaluated with SCC-DFTB only. Our findings further suggest that unlike the other stacking geometries modeled, serrated Zn<sub>3</sub>C<sub>6</sub>O<sub>6</sub>, serrated Zn-NH-MOF, and wavy serrated Ni<sub>3</sub>(HITP)<sub>2</sub> represent stable geometries. While Zn<sub>3</sub>C<sub>6</sub>O<sub>6</sub> and Zn-NH-MOF exhibit a higher power factor than Ni<sub>3</sub>(HITP)<sub>2</sub> (as found in our previous work), Zn-NH-MOF shows lower thermal conductivity, resulting in the highest thermoelectric figure of merit (<i>ZT</i>) among the studied MOFs.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"10167-10178"},"PeriodicalIF":5.5000,"publicationDate":"2024-11-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01229","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
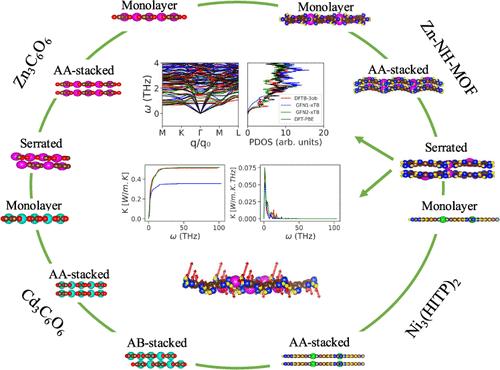
Despite the importance of modeling lattice thermal conductivity in predicting thermoelectric (TE) properties, computational data on heat transport, especially from first-principles, in 2D metal-organic frameworks (MOFs) remain limited due to the high computational cost. To address this, we provide a benchmark of the performance of semiempirical self-consistent-charge density functional tight-binding (SCC-DFTB) methods against density functional theory (DFT) for monolayer, serrated, AA-stacked and/or AB-stacked Zn3C6O6, Cd3C6O6, Zn-NH-MOF, and Ni3(HITP)2 MOFs. Harmonic lattice dynamics calculations, including partial atomic contributions to phonon dispersions, are evaluated with both SCC-DFTB and DFT, whereas anharmonic transport (i.e., thermal conductivity) is evaluated with SCC-DFTB only. Our findings further suggest that unlike the other stacking geometries modeled, serrated Zn3C6O6, serrated Zn-NH-MOF, and wavy serrated Ni3(HITP)2 represent stable geometries. While Zn3C6O6 and Zn-NH-MOF exhibit a higher power factor than Ni3(HITP)2 (as found in our previous work), Zn-NH-MOF shows lower thermal conductivity, resulting in the highest thermoelectric figure of merit (ZT) among the studied MOFs.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
