Wanjiang You , Hao Zou , Xiaoqiang Wang , Lielin Wang , Ning Pan , Fang Xiang
{"title":"First-principles density functional study of iodine molecule adsorption on stable CuS surfaces","authors":"Wanjiang You , Hao Zou , Xiaoqiang Wang , Lielin Wang , Ning Pan , Fang Xiang","doi":"10.1016/j.comptc.2024.114952","DOIUrl":null,"url":null,"abstract":"<div><div>The present work investigated the adsorption of gaseous iodine molecules (I<sub>2</sub>) on stable CuS surface, which has demonstrated excellent performance as an adsorbent for I<sub>2</sub> removal, with first-principles density functional theory (DFT). In this work, a pair of asymmetric surfaces (marked as slab1 and slab2) formed by breaking the weakest bond along (0<!--> <!-->0<!--> <!-->1) direction are chosen to present CuS surfaces. The findings indicate that the adsorption of I<sub>2</sub> molecules on the pristine CuS(0<!--> <!-->0<!--> <!-->1) surface is relatively weak, while surface defects significantly enhance the binding strength of I<sub>2</sub>. In particular, S-vacancy CuS(0<!--> <!-->0<!--> <!-->1) surfaces exhibit considerably higher adsorption energy for I<sub>2</sub> compared to Cu-vacancy surfaces. We found that the hollow and Cu-top sites are typically the dominant adsorption sites, and the initial orientation of I<sub>2</sub> relative to the surface also influences the adsorption performance.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114952"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004912","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/30 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
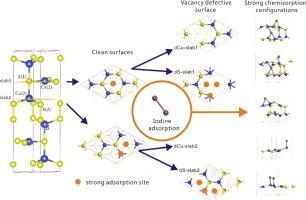
The present work investigated the adsorption of gaseous iodine molecules (I2) on stable CuS surface, which has demonstrated excellent performance as an adsorbent for I2 removal, with first-principles density functional theory (DFT). In this work, a pair of asymmetric surfaces (marked as slab1 and slab2) formed by breaking the weakest bond along (0 0 1) direction are chosen to present CuS surfaces. The findings indicate that the adsorption of I2 molecules on the pristine CuS(0 0 1) surface is relatively weak, while surface defects significantly enhance the binding strength of I2. In particular, S-vacancy CuS(0 0 1) surfaces exhibit considerably higher adsorption energy for I2 compared to Cu-vacancy surfaces. We found that the hollow and Cu-top sites are typically the dominant adsorption sites, and the initial orientation of I2 relative to the surface also influences the adsorption performance.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
