{"title":"Adsorption of Oxygen and Nitrogen Atoms on the SiO2 Surface: Molecular Dynamics Calculation Based on the Quantum-Mechanical Potential","authors":"A. A. Kroupnov, M. Ju. Pogosbekian","doi":"10.1134/S0015462824602675","DOIUrl":null,"url":null,"abstract":"<p>The processes of adsorption of N and O atoms on the surfaces of the thermal protection material SiO<sub>2</sub> are studied by the methods of quantum mechanics and molecular dynamics. The potential energy surface (PES) is calculated using the electron density functional theory. Based on the obtained PESs, the rate constants of adsorption of N and O atoms are determined by molecular dynamics methods in a wide range of surface temperatures of 500–2200 K and presented in the form of a generalized Arrhenius formula. The calculated rate constants are compared with the known phenomenological models and the calculation results based on the transition state theory.</p>","PeriodicalId":560,"journal":{"name":"Fluid Dynamics","volume":"59 4","pages":"924 - 931"},"PeriodicalIF":0.6000,"publicationDate":"2024-11-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Fluid Dynamics","FirstCategoryId":"5","ListUrlMain":"https://link.springer.com/article/10.1134/S0015462824602675","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"MECHANICS","Score":null,"Total":0}
引用次数: 0
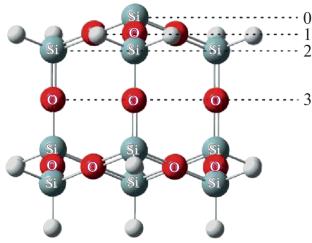
Abstract
The processes of adsorption of N and O atoms on the surfaces of the thermal protection material SiO2 are studied by the methods of quantum mechanics and molecular dynamics. The potential energy surface (PES) is calculated using the electron density functional theory. Based on the obtained PESs, the rate constants of adsorption of N and O atoms are determined by molecular dynamics methods in a wide range of surface temperatures of 500–2200 K and presented in the form of a generalized Arrhenius formula. The calculated rate constants are compared with the known phenomenological models and the calculation results based on the transition state theory.
本文采用量子力学和分子动力学方法研究了 N 原子和 O 原子在热防护材料 SiO2 表面的吸附过程。利用电子密度泛函理论计算了势能面(PES)。根据得到的势能面,用分子动力学方法确定了 N 原子和 O 原子在 500-2200 K 宽表面温度范围内的吸附速率常数,并以广义阿伦尼乌斯公式的形式表示出来。计算出的速率常数与已知的现象学模型和基于过渡态理论的计算结果进行了比较。
期刊介绍:
Fluid Dynamics is an international peer reviewed journal that publishes theoretical, computational, and experimental research on aeromechanics, hydrodynamics, plasma dynamics, underground hydrodynamics, and biomechanics of continuous media. Special attention is given to new trends developing at the leading edge of science, such as theory and application of multi-phase flows, chemically reactive flows, liquid and gas flows in electromagnetic fields, new hydrodynamical methods of increasing oil output, new approaches to the description of turbulent flows, etc.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
