{"title":"Uncertainty Qualification for Deep Learning-Based Elementary Reaction Property Prediction","authors":"Yan Liu, Yiming Mo* and Youwei Cheng*, ","doi":"10.1021/acs.jcim.4c0135810.1021/acs.jcim.4c01358","DOIUrl":null,"url":null,"abstract":"<p >The prediction of the thermodynamic and kinetic properties of elementary reactions has shown rapid improvement due to the implementation of deep learning (DL) methods. While various studies have reported the success in predicting reaction properties, the quantification of prediction uncertainty has seldom been investigated, thus compromising the confidence in using these predicted properties in practical applications. Here, we integrated graph convolutional neural networks (GCNN) with three uncertainty prediction techniques, including deep ensemble, Monte Carlo (MC)-dropout, and evidential learning, to provide insights into the uncertainty quantification and utility. The deep ensemble model outperforms others in accuracy and shows the highest reliability in estimating prediction uncertainty across all elementary reaction property data sets. We also verified that the deep ensemble model showed a satisfactory capability in recognizing epistemic and aleatoric uncertainties. Additionally, we adopted a Monte Carlo Tree Search method for extracting the explainable reaction substructures, providing a chemical explanation for DL predicted properties and corresponding uncertainties. Finally, to demonstrate the utility of uncertainty qualification in practical applications, we performed an uncertainty-guided calibration of the DL-constructed kinetic model, which achieved a 25% higher hit ratio in identifying dominant reaction pathways compared to that of the calibration without uncertainty guidance.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 21","pages":"8131–8141 8131–8141"},"PeriodicalIF":5.6000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c01358","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
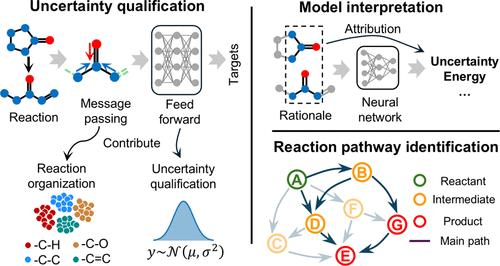
The prediction of the thermodynamic and kinetic properties of elementary reactions has shown rapid improvement due to the implementation of deep learning (DL) methods. While various studies have reported the success in predicting reaction properties, the quantification of prediction uncertainty has seldom been investigated, thus compromising the confidence in using these predicted properties in practical applications. Here, we integrated graph convolutional neural networks (GCNN) with three uncertainty prediction techniques, including deep ensemble, Monte Carlo (MC)-dropout, and evidential learning, to provide insights into the uncertainty quantification and utility. The deep ensemble model outperforms others in accuracy and shows the highest reliability in estimating prediction uncertainty across all elementary reaction property data sets. We also verified that the deep ensemble model showed a satisfactory capability in recognizing epistemic and aleatoric uncertainties. Additionally, we adopted a Monte Carlo Tree Search method for extracting the explainable reaction substructures, providing a chemical explanation for DL predicted properties and corresponding uncertainties. Finally, to demonstrate the utility of uncertainty qualification in practical applications, we performed an uncertainty-guided calibration of the DL-constructed kinetic model, which achieved a 25% higher hit ratio in identifying dominant reaction pathways compared to that of the calibration without uncertainty guidance.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
