Sustained applicability of SARS-CoV-2 variants identification by Sanger Sequencing Strategy on emerging various SARS-CoV-2 Omicron variants in Hiroshima, Japan.
{"title":"Sustained applicability of SARS-CoV-2 variants identification by Sanger Sequencing Strategy on emerging various SARS-CoV-2 Omicron variants in Hiroshima, Japan.","authors":"Chanroth Chhoung, Ko Ko, Serge Ouoba, Zayar Phyo, Golda Ataa Akuffo, Aya Sugiyama, Tomoyuki Akita, Hiroshi Sasaki, Tadashi Yamamoto, Kazuaki Takahashi, Junko Tanaka","doi":"10.1186/s12864-024-10973-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) persists, giving rise to new variants characterized by mutations in the spike protein. However, public data regarding the virus's evolutionary trend is not widely available after the downgrade of coronavirus disease 2019(COVID-19). Therefore, this study aimed to investigate the applicability of an in-house Sanger-based method for identifying SARS-CoV-2 variants, particularly focusing on newly emerged Omicron variants, and updating the epidemiology of COVID-19 during the 8<sup>th</sup> wave in Hiroshima Prefecture.</p><p><strong>Results: </strong>A total of 639 saliva samples of individuals who had tested positive for COVID-19, received from Hiroshima City Medical Association Clinical Laboratory Center between February 01, 2023, and March 12, 2024, were included in the study. SARS-CoV-2 variants were identified in 69.3% (443/639) with the mean viral titer 2 × 10<sup>6</sup> copies/mL, and high viral titer in Omicron variant XBC.1.6* (5 × 10<sup>8</sup> copies/mL) using RT-qPCR. By partial Spike gene-based sequencing using the Sanger Sequencing strategy, Omicron sub-lineages XXB.1, BA.5, and EG.1 were identified during different periods. A comprehensive phylogenetic analysis of 7383 SARS-CoV-2 strains retrieved from GISAID, collected in Hiroshima from the onset of the COVID-19 pandemic in early 2020 until July 2024, revealed the dynamic evolution of SARS-CoV-2 variants over time. The study found a similar pattern of variant distribution between the full genomes from GISAID, and the partial genomes obtained from our screening strategy during the same period.</p><p><strong>Conclusions: </strong>Our study revealed that all SARS-CoV-2 viruses circulated in Hiroshima were Omicron variants and their sub-lineages during the 8<sup>th</sup> wave outbreak in Hiroshima. Persistent molecular surveillance of SARS-CoV-2 is needed for the decision-making and strategic planning of the public promptly. Our study added evidence for the usefulness of SARS-CoV-2 spike gene partial sequencing-based SARS-CoV-2 variant identification strategy for mass screening and molecular surveillance even though the evolution of newly emerged various SARS-CoV-2 Omicron variants.</p>","PeriodicalId":9030,"journal":{"name":"BMC Genomics","volume":"25 1","pages":"1063"},"PeriodicalIF":3.7000,"publicationDate":"2024-11-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11552212/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12864-024-10973-0","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) persists, giving rise to new variants characterized by mutations in the spike protein. However, public data regarding the virus's evolutionary trend is not widely available after the downgrade of coronavirus disease 2019(COVID-19). Therefore, this study aimed to investigate the applicability of an in-house Sanger-based method for identifying SARS-CoV-2 variants, particularly focusing on newly emerged Omicron variants, and updating the epidemiology of COVID-19 during the 8th wave in Hiroshima Prefecture.
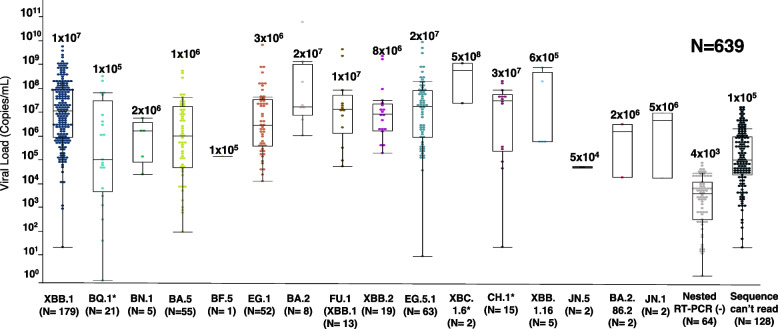
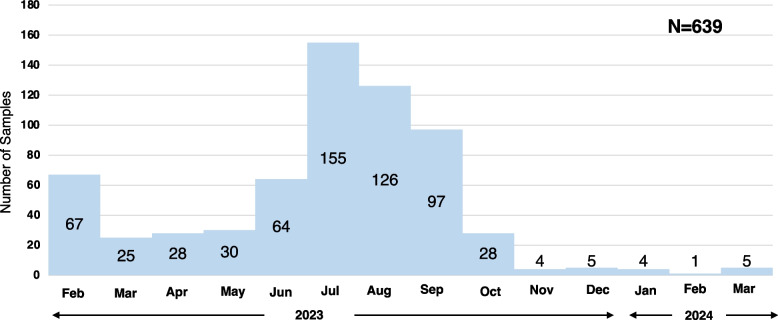
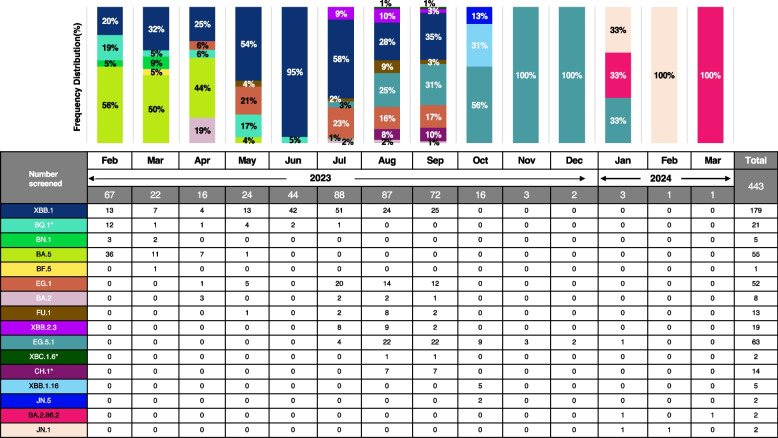
Results: A total of 639 saliva samples of individuals who had tested positive for COVID-19, received from Hiroshima City Medical Association Clinical Laboratory Center between February 01, 2023, and March 12, 2024, were included in the study. SARS-CoV-2 variants were identified in 69.3% (443/639) with the mean viral titer 2 × 106 copies/mL, and high viral titer in Omicron variant XBC.1.6* (5 × 108 copies/mL) using RT-qPCR. By partial Spike gene-based sequencing using the Sanger Sequencing strategy, Omicron sub-lineages XXB.1, BA.5, and EG.1 were identified during different periods. A comprehensive phylogenetic analysis of 7383 SARS-CoV-2 strains retrieved from GISAID, collected in Hiroshima from the onset of the COVID-19 pandemic in early 2020 until July 2024, revealed the dynamic evolution of SARS-CoV-2 variants over time. The study found a similar pattern of variant distribution between the full genomes from GISAID, and the partial genomes obtained from our screening strategy during the same period.
Conclusions: Our study revealed that all SARS-CoV-2 viruses circulated in Hiroshima were Omicron variants and their sub-lineages during the 8th wave outbreak in Hiroshima. Persistent molecular surveillance of SARS-CoV-2 is needed for the decision-making and strategic planning of the public promptly. Our study added evidence for the usefulness of SARS-CoV-2 spike gene partial sequencing-based SARS-CoV-2 variant identification strategy for mass screening and molecular surveillance even though the evolution of newly emerged various SARS-CoV-2 Omicron variants.
期刊介绍:
BMC Genomics is an open access, peer-reviewed journal that considers articles on all aspects of genome-scale analysis, functional genomics, and proteomics.
BMC Genomics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
