Jia Li, Patricia Vindel-Zandbergen, Jun Li*, Peter M. Felker* and Zlatko Bačić*,
{"title":"HF Trimer: A New Full-Dimensional Potential Energy Surface and Rigorous 12D Quantum Calculations of Vibrational States","authors":"Jia Li, Patricia Vindel-Zandbergen, Jun Li*, Peter M. Felker* and Zlatko Bačić*, ","doi":"10.1021/acs.jpca.4c0377110.1021/acs.jpca.4c03771","DOIUrl":null,"url":null,"abstract":"<p >HF trimer, as the smallest and the lightest cyclic hydrogen-bonded (HB) cluster, has long been a favorite prototype system for spectroscopic and theoretical investigations of the structure, energetics, spectroscopy, and dynamics of hydrogen-bond networks. Recently, rigorous quantum 12D calculations of the coupled intra- and intermolecular vibrations of this fundamental HB trimer (<i>J. Chem. Phys.</i> <b>2023</b>, <i>158</i>, 234109) were performed, employing an older ab initio-based many-body potential energy surface (PES). While the theoretical results were found to be in reasonably good agreement with the available spectroscopic data, it was also evident that it is highly desirable to develop a more accurate 12D PES of HF trimer. Motivated by this, here we report a new, and the first fully ab initio 12D PES of this paradigmatic system. Approximately 42,540 geometries were sampled and calculated at the level of CCSD(T)-F12a/AVTZ. The permutationally invariant polynomial-neural network based Δ-machine learning approach (<i>J. Phys. Chem. Lett.</i> <b>2022</b>, <i>13</i>, 4729) was employed to perform cost-efficient calculations of the basis-set-superposition error (BSSE) correction. By strategically selecting data points, this approach facilitated the construction of a high-precision PES with BSSE correction, while requiring only a minimal number of BSSE value computations. The fitting error of the final PES is only 0.035 kcal/mol. To assess its performance, the 12D fully coupled quantum calculations of excited intra- and intermolecular vibrational states of HF trimer are carried out using the rigorous methodology developed by us earlier. The results are found to be in a significantly better agreement with the available spectroscopic data than those obtained with the previously existing semiempirical 12D PES.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 45","pages":"9707–9720 9707–9720"},"PeriodicalIF":2.7000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c03771","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
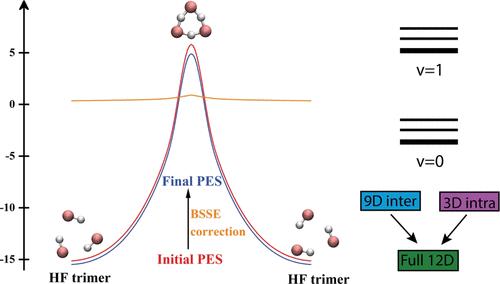
HF trimer, as the smallest and the lightest cyclic hydrogen-bonded (HB) cluster, has long been a favorite prototype system for spectroscopic and theoretical investigations of the structure, energetics, spectroscopy, and dynamics of hydrogen-bond networks. Recently, rigorous quantum 12D calculations of the coupled intra- and intermolecular vibrations of this fundamental HB trimer (J. Chem. Phys.2023, 158, 234109) were performed, employing an older ab initio-based many-body potential energy surface (PES). While the theoretical results were found to be in reasonably good agreement with the available spectroscopic data, it was also evident that it is highly desirable to develop a more accurate 12D PES of HF trimer. Motivated by this, here we report a new, and the first fully ab initio 12D PES of this paradigmatic system. Approximately 42,540 geometries were sampled and calculated at the level of CCSD(T)-F12a/AVTZ. The permutationally invariant polynomial-neural network based Δ-machine learning approach (J. Phys. Chem. Lett.2022, 13, 4729) was employed to perform cost-efficient calculations of the basis-set-superposition error (BSSE) correction. By strategically selecting data points, this approach facilitated the construction of a high-precision PES with BSSE correction, while requiring only a minimal number of BSSE value computations. The fitting error of the final PES is only 0.035 kcal/mol. To assess its performance, the 12D fully coupled quantum calculations of excited intra- and intermolecular vibrational states of HF trimer are carried out using the rigorous methodology developed by us earlier. The results are found to be in a significantly better agreement with the available spectroscopic data than those obtained with the previously existing semiempirical 12D PES.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
