Muzzakkir Amin, Mohammad Musfiqur Rahman, Md Kazi Rokunuzzaman, Md Kamal Hossain, Farid Ahmed
{"title":"First-principles study of aromatic amino acid encapsulation in single-walled BN and AlN nanotubes","authors":"Muzzakkir Amin, Mohammad Musfiqur Rahman, Md Kazi Rokunuzzaman, Md Kamal Hossain, Farid Ahmed","doi":"10.1016/j.comptc.2024.114954","DOIUrl":null,"url":null,"abstract":"<div><div>Aromatic molecules exhibit strong non-covalent interactions with nanotubes, influencing their encapsulation properties. This study uses DFT calculations to explore the encapsulation of aromatic amino acids within zigzag (ZZ), chiral (R/S), and armchair (AC) single-walled aluminum nitride nanotubes (AlNNTs) and boron nitride nanotubes (BNNTs). The results reveal that zigzag AlNNTs exhibit the highest encapsulation affinity compared to other chiralities, while chiral BNNTs show enhanced encapsulation. Encapsulation energy decreases with increasing nanotube radius, indicating reduced affinity. Overall, the studied BNNTs demonstrate stronger encapsulation energy compared to AlNNTs. The bandgap energy of the encapsulated structures varies significantly with nanotube diameter and chirality. The physisorption process plays a major role in encapsulation, affecting the geometric and electronic properties of the nanotubes and enhancing the stability and efficacy of the encapsulated amino acids. These findings highlight the potential of these nanostructures for advanced applications, including targeted drug delivery and molecular sensing.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114954"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004936","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/30 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
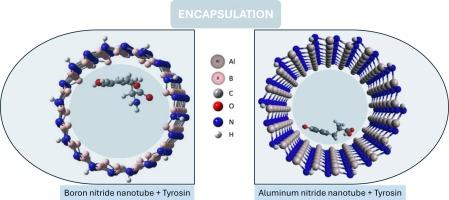
Aromatic molecules exhibit strong non-covalent interactions with nanotubes, influencing their encapsulation properties. This study uses DFT calculations to explore the encapsulation of aromatic amino acids within zigzag (ZZ), chiral (R/S), and armchair (AC) single-walled aluminum nitride nanotubes (AlNNTs) and boron nitride nanotubes (BNNTs). The results reveal that zigzag AlNNTs exhibit the highest encapsulation affinity compared to other chiralities, while chiral BNNTs show enhanced encapsulation. Encapsulation energy decreases with increasing nanotube radius, indicating reduced affinity. Overall, the studied BNNTs demonstrate stronger encapsulation energy compared to AlNNTs. The bandgap energy of the encapsulated structures varies significantly with nanotube diameter and chirality. The physisorption process plays a major role in encapsulation, affecting the geometric and electronic properties of the nanotubes and enhancing the stability and efficacy of the encapsulated amino acids. These findings highlight the potential of these nanostructures for advanced applications, including targeted drug delivery and molecular sensing.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
