{"title":"Enhanced performance of a promising Au/TMDC heterostructure composed of MoTe2 nanosheets decorated with Au5 clusters: A DFT study","authors":"Esmail Vessally , Rovnag Rzayev , Bayan Azizi , Pawan Sharma , Abhishek Kumar","doi":"10.1016/j.comptc.2024.114933","DOIUrl":null,"url":null,"abstract":"<div><div>This research uses density functional theory approach combined with the spin–orbit coupling to study how the SO<sub>x</sub> molecules stick to Au<sub>5</sub> cluster functionalized MoTe<sub>2</sub> nanosheets. In fact, the promising Au/MoTe<sub>2</sub> heterostructure system is constructed to model the attachment of gases on its surface. The high efficiency of adsorption process is evident from the strong sticking of the SO<sub>x</sub> to the Au atoms. Both Au<sub>1</sub> and Au<sub>5</sub> cluster modified MoTe<sub>2</sub> nanosheets revealed semiconducting feature, and in Au<sub>5</sub> cluster modified system, the band gap narrowed, while the conductivity is enhanced. Thus, results showed that adding Au cluster to the MoTe<sub>2</sub> made it best for adsorbing gases, while MoTe<sub>2</sub> without any additives absorbed the gases weakly. The conductivity and recovery time are also analyzed to further describe the results. Based on our theoretical consequences, the Au<sub>5</sub> cluster functionalized MoTe<sub>2</sub> (Au<sub>/</sub>MoTe<sub>2</sub> heterostructure system) seem good for constructing innovative sensors to detect SO<sub>x</sub> molecules.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114933"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004729","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/4 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
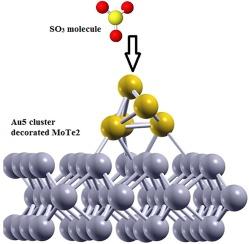
Abstract
This research uses density functional theory approach combined with the spin–orbit coupling to study how the SOx molecules stick to Au5 cluster functionalized MoTe2 nanosheets. In fact, the promising Au/MoTe2 heterostructure system is constructed to model the attachment of gases on its surface. The high efficiency of adsorption process is evident from the strong sticking of the SOx to the Au atoms. Both Au1 and Au5 cluster modified MoTe2 nanosheets revealed semiconducting feature, and in Au5 cluster modified system, the band gap narrowed, while the conductivity is enhanced. Thus, results showed that adding Au cluster to the MoTe2 made it best for adsorbing gases, while MoTe2 without any additives absorbed the gases weakly. The conductivity and recovery time are also analyzed to further describe the results. Based on our theoretical consequences, the Au5 cluster functionalized MoTe2 (Au/MoTe2 heterostructure system) seem good for constructing innovative sensors to detect SOx molecules.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
