How to get rid of imaginary frequencies within ONIOM geometry optimizations: A DFT study on the effect of basis set and link atom distances in Cu-ZSM-5
{"title":"How to get rid of imaginary frequencies within ONIOM geometry optimizations: A DFT study on the effect of basis set and link atom distances in Cu-ZSM-5","authors":"Michele De Rosa, Simone Morpurgo","doi":"10.1016/j.comptc.2024.114956","DOIUrl":null,"url":null,"abstract":"<div><div>Two extended clusters representing different portions of Cu-ZSM-5 were treated within a two-layer ONIOM approximation, employing DFT calculations for both the real and the model system. Despite a two-step optimization procedure successfully employed in previous work, a consistent number of imaginary and anomalous frequencies appeared after the vibrational analysis. These artefacts depend both on the basis set assigned to link atoms and on an improper setting of the O–H distances, where H are the link atoms at the boundaries of the model system. The latter problem, revealed for the first time in the present study, originates from the default scale factor employed by the ONIOM routine within Gaussian-09. Once basis set and <em>g</em> scale factor are properly set, all imaginary and anomalous frequencies disappear. The present findings may represent an interesting and practical solution to an annoying computational problem, whenever it occurs in the framework of ONIOM calculations.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114956"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X2400495X","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/1 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
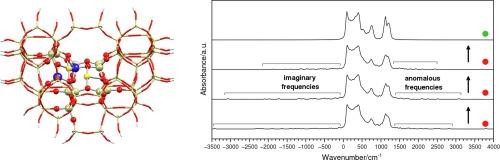
Two extended clusters representing different portions of Cu-ZSM-5 were treated within a two-layer ONIOM approximation, employing DFT calculations for both the real and the model system. Despite a two-step optimization procedure successfully employed in previous work, a consistent number of imaginary and anomalous frequencies appeared after the vibrational analysis. These artefacts depend both on the basis set assigned to link atoms and on an improper setting of the O–H distances, where H are the link atoms at the boundaries of the model system. The latter problem, revealed for the first time in the present study, originates from the default scale factor employed by the ONIOM routine within Gaussian-09. Once basis set and g scale factor are properly set, all imaginary and anomalous frequencies disappear. The present findings may represent an interesting and practical solution to an annoying computational problem, whenever it occurs in the framework of ONIOM calculations.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
