Arianna Massaro, Francesca Fasulo, Ana B. Muñoz-García, Michele Pavone
{"title":"Elucidating Structural and Electronic Features of PEO/(101)-TiO2 Anatase Interfaces through First-Principles Metadynamics Simulations","authors":"Arianna Massaro, Francesca Fasulo, Ana B. Muñoz-García, Michele Pavone","doi":"10.1021/acs.jpcc.4c07016","DOIUrl":null,"url":null,"abstract":"Structural and dynamic properties of the poly(ethylene oxide)/anatase interface (<i>i.e.</i>, PEO/TiO<sub>2</sub>) are explored <i>via</i> metadynamics simulations and density functional tight binding. 1-ns trajectories are reconstructed upon two different structure-related collective variables: interfacial Ti<sub>surf</sub>–O<sub>PEO</sub> distances and polymer chain torsion angles. The conformational freedom of PEO is significantly influenced by multiple favorable interactions with unsaturated Ti sites on the anatase surface. From these trajectories, several equilibrium structures extracted from the free-energy surface are analyzed using electronic structure calculations within density functional theory: the titania work function results in being largely influenced by the dynamically resolved structuring of PEO on the anatase surface. Besides the intrinsic valuable insights into the technologically relevant PEO/TiO<sub>2</sub> interface, our findings provide the first innovative example of an affordable yet reliable computational protocol to describe a heterogeneous interface and predict the effects of molecular dynamics on relevant physicochemical properties.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"249 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-11-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c07016","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
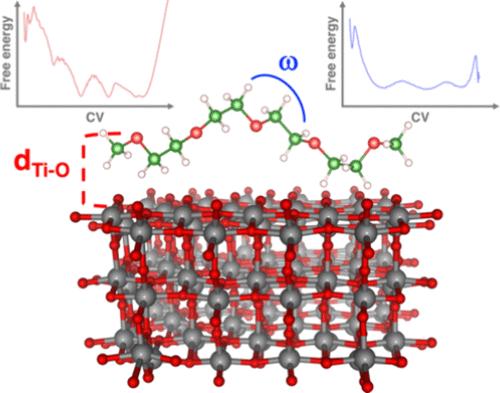
Structural and dynamic properties of the poly(ethylene oxide)/anatase interface (i.e., PEO/TiO2) are explored via metadynamics simulations and density functional tight binding. 1-ns trajectories are reconstructed upon two different structure-related collective variables: interfacial Tisurf–OPEO distances and polymer chain torsion angles. The conformational freedom of PEO is significantly influenced by multiple favorable interactions with unsaturated Ti sites on the anatase surface. From these trajectories, several equilibrium structures extracted from the free-energy surface are analyzed using electronic structure calculations within density functional theory: the titania work function results in being largely influenced by the dynamically resolved structuring of PEO on the anatase surface. Besides the intrinsic valuable insights into the technologically relevant PEO/TiO2 interface, our findings provide the first innovative example of an affordable yet reliable computational protocol to describe a heterogeneous interface and predict the effects of molecular dynamics on relevant physicochemical properties.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
