Chengchun Zhu , Leilei Li , Yan Yu , Xiao Wang , Ying Shi , Yiping Gao , Kai Chen , Xiaoyu Liu , Yuqian Cui , Tao Zhang , Zhiyi Yu
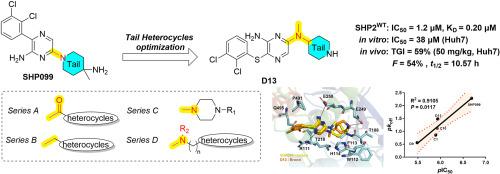
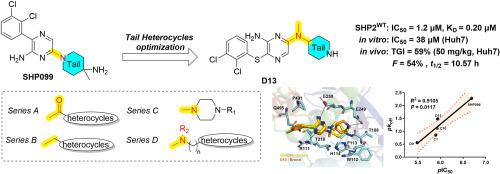
{"title":"Optimization of SHP2 allosteric inhibitors with novel tail heterocycles and their potential as antitumor therapeutics","authors":"Chengchun Zhu , Leilei Li , Yan Yu , Xiao Wang , Ying Shi , Yiping Gao , Kai Chen , Xiaoyu Liu , Yuqian Cui , Tao Zhang , Zhiyi Yu","doi":"10.1016/j.ejmech.2024.117078","DOIUrl":null,"url":null,"abstract":"<div><div>SHP2, a non-receptor protein tyrosine phosphatase involved in cancers, plays a pivotal role in numerous cellular signaling cascades, including the MAPK and PD-L1/PD-1 pathways. Although several SHP2 allosteric inhibitors have already entered clinical trials, none have been approved to date. Therefore, the development of new SHP2 allosteric inhibitors with improved efficacy is urgently required. Herein, we report the optimization of tail heterocycles in SHP2 allosteric inhibitors using a structure-based drug design strategy. Four series of compounds with different tail skeletons were synthesized, among which <strong>D13</strong> showed notable inhibitory activity (IC<sub>50</sub> = 1.2 μM) against SHP2. Molecular docking and binding studies indicated that the newly synthesized compounds exerted enzymatic inhibitory effects by directly binding to SHP2 with relatively slow dissociation rates. At the cellular level, Huh7 cells demonstrated heightened sensitivity to the novel SHP2 inhibitors, and <strong>D13</strong> exhibited superior antiproliferative activity (IC<sub>50</sub> = 38 μM) by arresting G0/G1 cell cycle, facilitating cell apoptosis and suppressing the MAPK signaling pathway. In the <em>in vivo</em> study, <strong>D13</strong> displayed significant antitumor activity in a Huh7 xenograft model and possessed favorable druggability with acceptable oral bioavailability (<em>F</em> = 54 %) and half-life (<em>t</em><sub>1/2</sub> = 10.57 h). Collectively, this study lays a robust foundation for further optimization of the tail heterocycle skeleton in SHP2 allosteric inhibitors.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"282 ","pages":"Article 117078"},"PeriodicalIF":5.9000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0223523424009607","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/18 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
SHP2, a non-receptor protein tyrosine phosphatase involved in cancers, plays a pivotal role in numerous cellular signaling cascades, including the MAPK and PD-L1/PD-1 pathways. Although several SHP2 allosteric inhibitors have already entered clinical trials, none have been approved to date. Therefore, the development of new SHP2 allosteric inhibitors with improved efficacy is urgently required. Herein, we report the optimization of tail heterocycles in SHP2 allosteric inhibitors using a structure-based drug design strategy. Four series of compounds with different tail skeletons were synthesized, among which D13 showed notable inhibitory activity (IC50 = 1.2 μM) against SHP2. Molecular docking and binding studies indicated that the newly synthesized compounds exerted enzymatic inhibitory effects by directly binding to SHP2 with relatively slow dissociation rates. At the cellular level, Huh7 cells demonstrated heightened sensitivity to the novel SHP2 inhibitors, and D13 exhibited superior antiproliferative activity (IC50 = 38 μM) by arresting G0/G1 cell cycle, facilitating cell apoptosis and suppressing the MAPK signaling pathway. In the in vivo study, D13 displayed significant antitumor activity in a Huh7 xenograft model and possessed favorable druggability with acceptable oral bioavailability (F = 54 %) and half-life (t1/2 = 10.57 h). Collectively, this study lays a robust foundation for further optimization of the tail heterocycle skeleton in SHP2 allosteric inhibitors.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
