Nobuyuki N. Matsuzawa, Hiroyuki Maeshima, Keisuke Hayashi, Tatsuhito Ando, Mohammad Atif Faiz Afzal, Kyle Marshall, Benjamin J. Coscia, Andrea R. Browning, Alexander Goldberg, Mathew D. Halls, Karl Leswing, Mayank Misra, Farhad Ramezanghorbani, Tsuguo Morisato
{"title":"Exploring Molecules with Low Viscosity: Using Physics-Based Simulations and De Novo Design by Applying Reinforcement Learning","authors":"Nobuyuki N. Matsuzawa, Hiroyuki Maeshima, Keisuke Hayashi, Tatsuhito Ando, Mohammad Atif Faiz Afzal, Kyle Marshall, Benjamin J. Coscia, Andrea R. Browning, Alexander Goldberg, Mathew D. Halls, Karl Leswing, Mayank Misra, Farhad Ramezanghorbani, Tsuguo Morisato","doi":"10.1021/acs.chemmater.4c02929","DOIUrl":null,"url":null,"abstract":"Molecules with viscosities lower than those of conventional organic solvents are highly sought after for applications in electrochemical devices such as batteries and capacitors. These molecules improve the electrical resistance of devices, enhancing their efficiency, especially at low temperatures. To identify new molecules with low viscosities, we conducted extensive molecular dynamics (MD) simulations on 10,000 molecules selected from the GDB-17 chemical structure database, specifically choosing molecules with fewer than 12 heavy atoms. Additionally, we performed density functional theory (DFT) calculations to determine the energies of the highest occupied molecular orbitals (HOMO) of these molecules as a surrogate for the oxidation potential. We used the data on viscosity and HOMO levels as training sets to develop machine-learning models that predict these properties. Using these models, we carried out molecular de novo design using the REINVENT method, a reinforcement-learning approach utilizing SMILES strings. This method aimed to identify molecules that minimize viscosity while maintaining sufficiently low HOMO levels for stability. The approach successfully identified new chemical structures with viscosities below 2 mPa·s and suitably low HOMO energies. We synthesized a novel compound from the top candidates and validated our predictions experimentally. The experimental results closely matched our predictions, demonstrating that combining physics-based simulations with reinforcement learning is an effective strategy for designing novel molecules with targeted properties.","PeriodicalId":33,"journal":{"name":"Chemistry of Materials","volume":"226 1","pages":""},"PeriodicalIF":7.0000,"publicationDate":"2024-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemistry of Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1021/acs.chemmater.4c02929","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
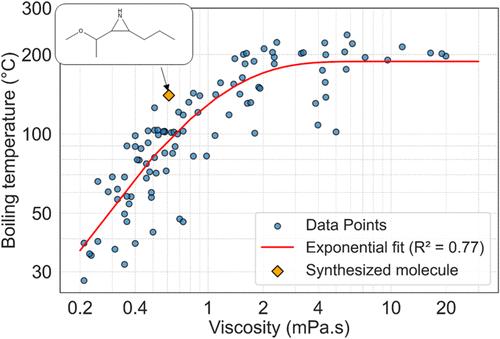
Molecules with viscosities lower than those of conventional organic solvents are highly sought after for applications in electrochemical devices such as batteries and capacitors. These molecules improve the electrical resistance of devices, enhancing their efficiency, especially at low temperatures. To identify new molecules with low viscosities, we conducted extensive molecular dynamics (MD) simulations on 10,000 molecules selected from the GDB-17 chemical structure database, specifically choosing molecules with fewer than 12 heavy atoms. Additionally, we performed density functional theory (DFT) calculations to determine the energies of the highest occupied molecular orbitals (HOMO) of these molecules as a surrogate for the oxidation potential. We used the data on viscosity and HOMO levels as training sets to develop machine-learning models that predict these properties. Using these models, we carried out molecular de novo design using the REINVENT method, a reinforcement-learning approach utilizing SMILES strings. This method aimed to identify molecules that minimize viscosity while maintaining sufficiently low HOMO levels for stability. The approach successfully identified new chemical structures with viscosities below 2 mPa·s and suitably low HOMO energies. We synthesized a novel compound from the top candidates and validated our predictions experimentally. The experimental results closely matched our predictions, demonstrating that combining physics-based simulations with reinforcement learning is an effective strategy for designing novel molecules with targeted properties.
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
