{"title":"Accelerating Molecular Dynamics Simulations Using Socket-Based Interprocess Communication","authors":"Matheus de Oliveira Bispo, Mario Barbatti","doi":"10.1021/acs.jpclett.4c02860","DOIUrl":null,"url":null,"abstract":"Molecular dynamics (MD) simulations are essential for studying the time evolution of molecular systems. Still, their efficiency is often bottlenecked by file-based interprocess communication (IPC) between MD and electronic structure programs. We present a socket-based IPC implementation that dramatically accelerates MD simulations, reducing the computational time by >10-fold compared to those of traditional file-based methods. Our approach, applied to nonadiabatic molecular dynamics with the Newton-X program, eliminates disk read/write overhead, allowing for faster simulations over longer time scales. This method opens the door to more efficient high-throughput simulations, providing new opportunities for exploring complex molecular processes in real time.","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"193 1","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2024-11-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c02860","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
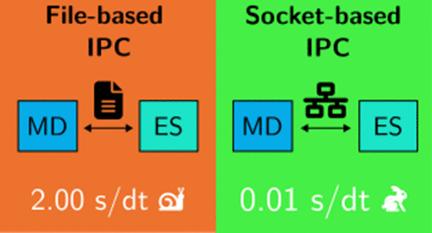
Molecular dynamics (MD) simulations are essential for studying the time evolution of molecular systems. Still, their efficiency is often bottlenecked by file-based interprocess communication (IPC) between MD and electronic structure programs. We present a socket-based IPC implementation that dramatically accelerates MD simulations, reducing the computational time by >10-fold compared to those of traditional file-based methods. Our approach, applied to nonadiabatic molecular dynamics with the Newton-X program, eliminates disk read/write overhead, allowing for faster simulations over longer time scales. This method opens the door to more efficient high-throughput simulations, providing new opportunities for exploring complex molecular processes in real time.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
