{"title":"Choice of Layering and Band Alignment in 2D Heterostructures","authors":"Raheel Hammad, Snehith Adabala, Soumya Ghosh","doi":"10.1021/acs.jpcc.4c06027","DOIUrl":null,"url":null,"abstract":"Heterostructures are ubiquitous in many optoelectronic devices and as photocatalysts. One of the key features of a heterojunction is proper band alignment between the two materials. Estimation of the correct relative band positions with density functional theory (DFT)-based electronic structure calculations is often constrained by the accuracy and cost associated with the various DFT functionals. In this study, we introduce a novel computational approach that achieves band alignments closely matching experimental results with the widely used PBE functional. We specifically examine the well-documented MoO<sub>3</sub>/MoS<sub>2</sub> system, a type-II heterojunction. In our setup, the MoS<sub>2</sub> layers are kept as they are, but for MoO<sub>3</sub>, the individual layers are chosen differently. These alternative layers have higher surface energy, and hence, the band edges are higher than those of the conventional layers. This shift in band edges of the alternative MoO<sub>3</sub> layers changes the band alignment in the MoO<sub>3</sub>/MoS<sub>2</sub> heterojunction from type-III to the experimentally observed type-II character. We also extend this computational strategy to additional systems, demonstrating its versatility and effectiveness.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"7 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-11-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c06027","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
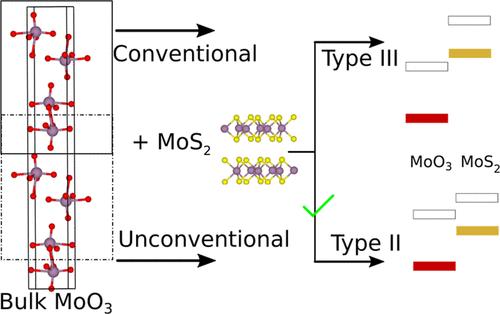
Heterostructures are ubiquitous in many optoelectronic devices and as photocatalysts. One of the key features of a heterojunction is proper band alignment between the two materials. Estimation of the correct relative band positions with density functional theory (DFT)-based electronic structure calculations is often constrained by the accuracy and cost associated with the various DFT functionals. In this study, we introduce a novel computational approach that achieves band alignments closely matching experimental results with the widely used PBE functional. We specifically examine the well-documented MoO3/MoS2 system, a type-II heterojunction. In our setup, the MoS2 layers are kept as they are, but for MoO3, the individual layers are chosen differently. These alternative layers have higher surface energy, and hence, the band edges are higher than those of the conventional layers. This shift in band edges of the alternative MoO3 layers changes the band alignment in the MoO3/MoS2 heterojunction from type-III to the experimentally observed type-II character. We also extend this computational strategy to additional systems, demonstrating its versatility and effectiveness.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
