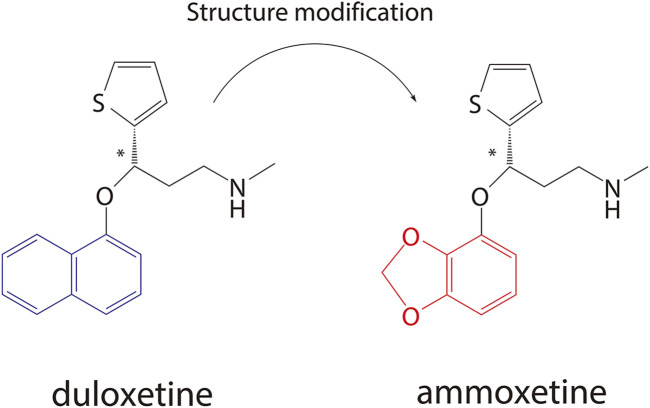
In vitro, in vivo, and in silico approaches for evaluating the preclinical DMPK profiles of ammoxetine, a novel chiral serotonin and norepinephrine reuptake inhibitor.
{"title":"<i>In vitro</i>, <i>in vivo</i>, and <i>in silico</i> approaches for evaluating the preclinical DMPK profiles of ammoxetine, a novel chiral serotonin and norepinephrine reuptake inhibitor.","authors":"Xiuqing Zhu, Yuexin Li, Huan Luo, Yunxia Zhang, Zhenqing Zhang, Jinglai Li","doi":"10.3389/fphar.2024.1486856","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and aim: </strong>Ammoxetine, a novel chiral serotonin and norepinephrine reuptake inhibitor, holds promise for major depressive disorder treatment. This study aimed to thoroughly investigate its preclinical drug metabolism and pharmacokinetics (DMPK) profiles.</p><p><strong>Methods: </strong>The preclinical DMPK profiles of ammoxetine were examined through <i>in vitro</i>, <i>in vivo</i>, and <i>in silico</i> methods.</p><p><strong>Results: </strong>Assessment of blood-brain barrier penetration <i>via</i> MDCK-MDR1 cells revealed strong brain permeation by ammoxetine, despite being a probable P-glycoprotein (P-gp) substrate. Molecular docking indicated a robust binding interaction between ammoxetine and P-gp. Ammoxetine was well absorbed orally, with T<sub>max</sub> ranging from 0.75 to 3.83 h in rats and 0.75-1.40 h in beagle dogs. At a 2 mg/kg dose in beagle dogs, ammoxetine exhibited an absolute bioavailability of approximately 42%. Plasma protein binding rates were around 50%-60% in beagle dogs, rats, and humans, suggesting moderate binding. Tissue distribution studies displayed rapid and extensive ammoxetine spread in major rat tissues post-gavage, with notable brain exposure and no tissue accumulation. Cumulative excretion rates in rats' urine, feces, and bile accounted for only 1.11% of the total administered drug, indicating extensive transformation into metabolites. Chiral inversion of ammoxetine was absent <i>in vivo</i>. Metabolic stability varied across species using liver microsomes, but beagle dogs showed clearance rates more akin to humans. Metabolic pathways unveiled two key metabolites, M1 and M2. M1, likely generated through methylenedioxyphenyl ring oxidation, involves CYP2C19 and CYP3A4, crucial human cytochrome P450 (CYP) enzymes for liver metabolism, while M2 is M1's glucuronide conjugate. Ammoxetine may exhibit saturation elimination trends with increasing doses in rats and beagle dogs. A high-throughput assay using the cocktail-substrate method indicated weak CYP inhibition by ammoxetine on CYP2D6 and CYP1A2, with minimal effects on other CYP enzymes, suggesting a low likelihood of CYP inhibition-related drug-drug interactions.</p><p><strong>Conclusion: </strong>This study presents encouraging DMPK profiles of ammoxetine, backing its potential as a candidate compound for future clinical assessments.</p>","PeriodicalId":12491,"journal":{"name":"Frontiers in Pharmacology","volume":"15 ","pages":"1486856"},"PeriodicalIF":4.8000,"publicationDate":"2024-11-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11579541/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Pharmacology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3389/fphar.2024.1486856","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and aim: Ammoxetine, a novel chiral serotonin and norepinephrine reuptake inhibitor, holds promise for major depressive disorder treatment. This study aimed to thoroughly investigate its preclinical drug metabolism and pharmacokinetics (DMPK) profiles.
Methods: The preclinical DMPK profiles of ammoxetine were examined through in vitro, in vivo, and in silico methods.
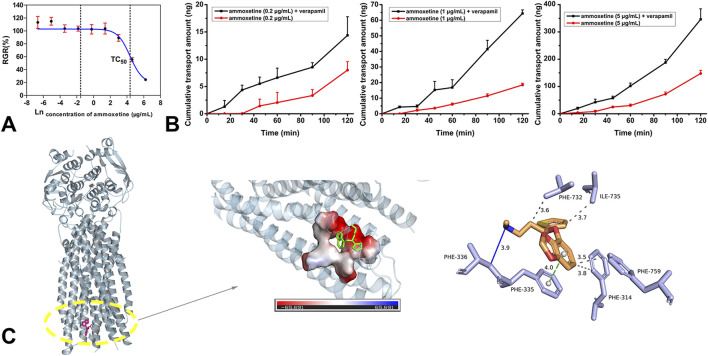
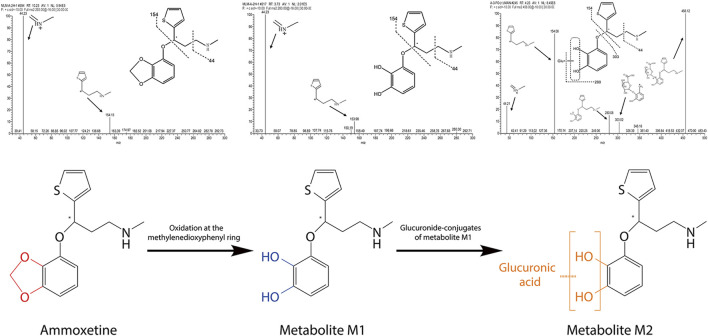
Results: Assessment of blood-brain barrier penetration via MDCK-MDR1 cells revealed strong brain permeation by ammoxetine, despite being a probable P-glycoprotein (P-gp) substrate. Molecular docking indicated a robust binding interaction between ammoxetine and P-gp. Ammoxetine was well absorbed orally, with Tmax ranging from 0.75 to 3.83 h in rats and 0.75-1.40 h in beagle dogs. At a 2 mg/kg dose in beagle dogs, ammoxetine exhibited an absolute bioavailability of approximately 42%. Plasma protein binding rates were around 50%-60% in beagle dogs, rats, and humans, suggesting moderate binding. Tissue distribution studies displayed rapid and extensive ammoxetine spread in major rat tissues post-gavage, with notable brain exposure and no tissue accumulation. Cumulative excretion rates in rats' urine, feces, and bile accounted for only 1.11% of the total administered drug, indicating extensive transformation into metabolites. Chiral inversion of ammoxetine was absent in vivo. Metabolic stability varied across species using liver microsomes, but beagle dogs showed clearance rates more akin to humans. Metabolic pathways unveiled two key metabolites, M1 and M2. M1, likely generated through methylenedioxyphenyl ring oxidation, involves CYP2C19 and CYP3A4, crucial human cytochrome P450 (CYP) enzymes for liver metabolism, while M2 is M1's glucuronide conjugate. Ammoxetine may exhibit saturation elimination trends with increasing doses in rats and beagle dogs. A high-throughput assay using the cocktail-substrate method indicated weak CYP inhibition by ammoxetine on CYP2D6 and CYP1A2, with minimal effects on other CYP enzymes, suggesting a low likelihood of CYP inhibition-related drug-drug interactions.
Conclusion: This study presents encouraging DMPK profiles of ammoxetine, backing its potential as a candidate compound for future clinical assessments.
期刊介绍:
Frontiers in Pharmacology is a leading journal in its field, publishing rigorously peer-reviewed research across disciplines, including basic and clinical pharmacology, medicinal chemistry, pharmacy and toxicology. Field Chief Editor Heike Wulff at UC Davis is supported by an outstanding Editorial Board of international researchers. This multidisciplinary open-access journal is at the forefront of disseminating and communicating scientific knowledge and impactful discoveries to researchers, academics, clinicians and the public worldwide.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
