Vu Tien Lam , Nguyen Huu Lam , Duong Quoc Van , Nguyen Hoang Thoan , Nguyen Ngoc Trung , Dang Duc Dung
{"title":"Role of substitution Zr-site on the electronic structure and magnetic properties of BaTiO3 materials: A first principles calculation","authors":"Vu Tien Lam , Nguyen Huu Lam , Duong Quoc Van , Nguyen Hoang Thoan , Nguyen Ngoc Trung , Dang Duc Dung","doi":"10.1016/j.comptc.2024.114989","DOIUrl":null,"url":null,"abstract":"<div><div>In this study, we employ density functional theory calculations to investigate the electronic band structures, density of states, and magnetic properties of pure BaTiO<sub>3</sub> and Zr-doped BaTiO<sub>3</sub> compounds. The intrinsic BaTiO<sub>3</sub> compound exhibits an indirect band gap of 1.80 eV, which is larger than the range of 1.61–1.71 eV observed for Zr-doped BaTiO<sub>3</sub>. By systematically exploring the Zr substitution at different Ti sites, we analyze the impact of Zr doping on the electronic structure of BaTiO<sub>3</sub> materials. The introduction of Zr doping induces significant modifications in the band structure and density of states, resulting in the emergence of new energy levels within the band gap. These findings enhance the understanding of the electronic properties of BaTiO<sub>3</sub> and Zr-doped BaTiO<sub>3</sub> materials, paving the way for potential applications in electronic and optical devices.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1243 ","pages":"Article 114989"},"PeriodicalIF":3.0000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24005280","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/13 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
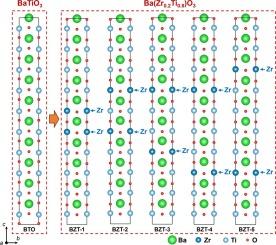
In this study, we employ density functional theory calculations to investigate the electronic band structures, density of states, and magnetic properties of pure BaTiO3 and Zr-doped BaTiO3 compounds. The intrinsic BaTiO3 compound exhibits an indirect band gap of 1.80 eV, which is larger than the range of 1.61–1.71 eV observed for Zr-doped BaTiO3. By systematically exploring the Zr substitution at different Ti sites, we analyze the impact of Zr doping on the electronic structure of BaTiO3 materials. The introduction of Zr doping induces significant modifications in the band structure and density of states, resulting in the emergence of new energy levels within the band gap. These findings enhance the understanding of the electronic properties of BaTiO3 and Zr-doped BaTiO3 materials, paving the way for potential applications in electronic and optical devices.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
