Cătălin Vasile Munteanu, Cătălin Marian, Adela Chiriță-Emandi, Maria Puiu, Adrian Pavel Trifa
{"title":"In silico splicing analysis of the PMS2 gene: exploring alternative molecular mechanisms in PMS2-associated Lynch syndrome.","authors":"Cătălin Vasile Munteanu, Cătălin Marian, Adela Chiriță-Emandi, Maria Puiu, Adrian Pavel Trifa","doi":"10.1186/s12863-024-01281-3","DOIUrl":null,"url":null,"abstract":"<p><p>Lynch syndrome (LS) is one of the most common hereditary cancer syndrome in human populations, associated with germline variants in MLH1, MSH2/EPCAM, MSH6 and PMS2 genes. The advent of next generation sequencing has proven a significant impact in germline variant detection in the causative genes; however, a large proportion of patients with clinical criteria still receive uncertain or negative results. PMS2 is the least frequent reported gene, associated with up to 15% of LS cases with late-onset disease and low penetrance phenotype; however, the proportion of PMS2-LS cases is considered to be highly underestimated. In this context, our analysis aimed to improve the current diagnostic yield by focusing on missense and intronic PMS2 variants available in public clinical databases (ClinVar, LOVD). We performed an in silico assessment of the wild-type DNA sequence and the reported genetic variants, employing splicing bioinformatics tools known for their effectiveness in other genes. Splicing variants were predicted in silico and using GTEx short-read RNA expression data. Out of the 2384 missense variants discovered, 90% were classified with uncertain significance (VUS). 4.9% of missense variants were shown to have a potential splicing consequence (DS > 0.2) using SpliceAI. As described in the original publication, SpliceAI-visual was proven effective in annotation of short intronic variants (< 50 bp). Four short intronic variants were identified using SpliceAI-visual as potentially splicing disturbing, in spite of using a lower threshold (DS > 0.1). Exons 2, 3, 4, 5, 6, 7, 8, 11, 12 and 14 were consistently predicted in at least three out of eight software with weak canonical splice sites. Additionally, we noted that both Exonic Splicing Enhancers (ESEs) and Exonic Splicing Silencers (ESSs) contribute significantly to alternative splicing and exonic selection in PMS2 gene. Specifically, ESE motifs were consistently more abundant in highly expressed exons 5, 11 and 14, while ESS motifs played a fundamental role in exons 6, 7 and 10. Computational analysis performed in our study serves as a valuable filtering step for guiding further RNA experiments. Additional functional data is necessary to validate our findings.</p>","PeriodicalId":72427,"journal":{"name":"BMC genomic data","volume":"25 1","pages":"100"},"PeriodicalIF":2.5000,"publicationDate":"2024-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11600730/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC genomic data","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12863-024-01281-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
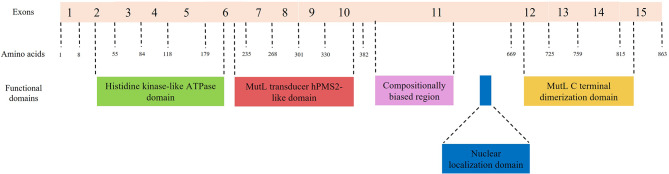
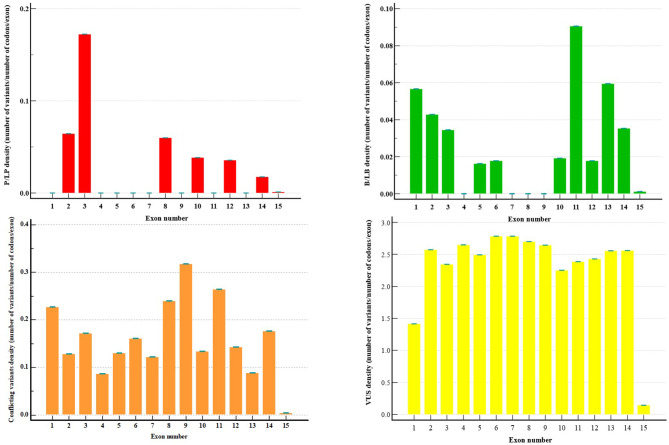
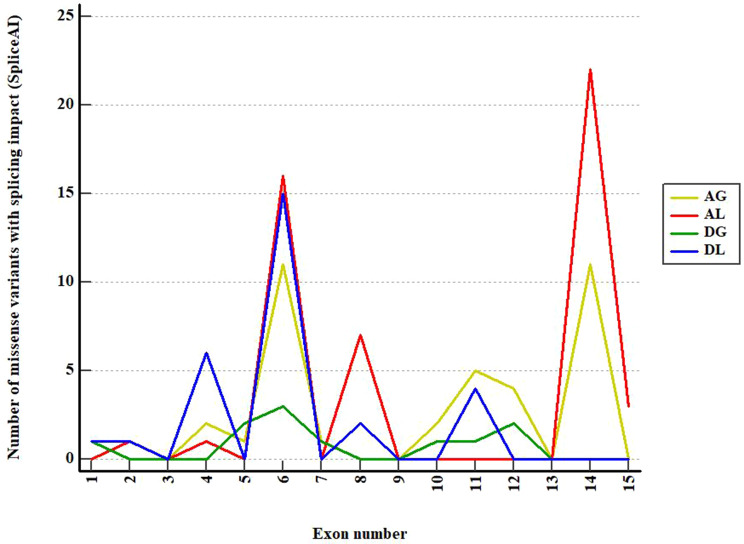
Lynch syndrome (LS) is one of the most common hereditary cancer syndrome in human populations, associated with germline variants in MLH1, MSH2/EPCAM, MSH6 and PMS2 genes. The advent of next generation sequencing has proven a significant impact in germline variant detection in the causative genes; however, a large proportion of patients with clinical criteria still receive uncertain or negative results. PMS2 is the least frequent reported gene, associated with up to 15% of LS cases with late-onset disease and low penetrance phenotype; however, the proportion of PMS2-LS cases is considered to be highly underestimated. In this context, our analysis aimed to improve the current diagnostic yield by focusing on missense and intronic PMS2 variants available in public clinical databases (ClinVar, LOVD). We performed an in silico assessment of the wild-type DNA sequence and the reported genetic variants, employing splicing bioinformatics tools known for their effectiveness in other genes. Splicing variants were predicted in silico and using GTEx short-read RNA expression data. Out of the 2384 missense variants discovered, 90% were classified with uncertain significance (VUS). 4.9% of missense variants were shown to have a potential splicing consequence (DS > 0.2) using SpliceAI. As described in the original publication, SpliceAI-visual was proven effective in annotation of short intronic variants (< 50 bp). Four short intronic variants were identified using SpliceAI-visual as potentially splicing disturbing, in spite of using a lower threshold (DS > 0.1). Exons 2, 3, 4, 5, 6, 7, 8, 11, 12 and 14 were consistently predicted in at least three out of eight software with weak canonical splice sites. Additionally, we noted that both Exonic Splicing Enhancers (ESEs) and Exonic Splicing Silencers (ESSs) contribute significantly to alternative splicing and exonic selection in PMS2 gene. Specifically, ESE motifs were consistently more abundant in highly expressed exons 5, 11 and 14, while ESS motifs played a fundamental role in exons 6, 7 and 10. Computational analysis performed in our study serves as a valuable filtering step for guiding further RNA experiments. Additional functional data is necessary to validate our findings.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
