{"title":"Pressure-induced structural and electronic properties of inorganic halide perovskite CsPbBr3","authors":"Zhixiang Geng, Shimin Chen, Chenhao Shang, Limin Chen, Chunsheng Liu, Qiyun Xie","doi":"10.1016/j.jssc.2024.125111","DOIUrl":null,"url":null,"abstract":"<div><div>In this paper, Density Functional Theory (DFT) based on first principles is used to study the influence of pressure on the structural and electronic properties of the cubic and orthorhombic phases of halogen perovskite CsPbBr<sub>3</sub>. We observe that the lattice constant of the cubic phase shows a monotonically decreasing trend in a wide pressure range, while the Pb–Br irregular octahedron of the orthorhombic phase rotates under pressure and distorts at 65.5 GPa, 67 GPa, 69.5 GPa, and 70 GPa pressure points, respectively. Moreover, the pressure has a good adjustment effect on the band gap of the cubic and orthorhombic phases. The band gap of the orthorhombic phase changes between the direct band gap and the indirect band gap under pressure. The charge difference density and density of states are calculated to study the type of bonds between internal atoms and the effect of pressure on the distribution of orbital electrons. The study of the Crystal Orbital Hamilton Populations and lattice energy of the electron cloud shows that the pressure has a significant adjustment effect on the strength of the Pb–Br covalent bond and the Cs–Br ionic bond. These results provide a comprehensive reference and guidance for the further research and application of perovskite material pressure engineering.</div></div>","PeriodicalId":378,"journal":{"name":"Journal of Solid State Chemistry","volume":"342 ","pages":"Article 125111"},"PeriodicalIF":3.5000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Solid State Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022459624005656","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/22 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
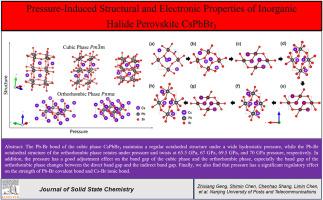
Abstract
In this paper, Density Functional Theory (DFT) based on first principles is used to study the influence of pressure on the structural and electronic properties of the cubic and orthorhombic phases of halogen perovskite CsPbBr3. We observe that the lattice constant of the cubic phase shows a monotonically decreasing trend in a wide pressure range, while the Pb–Br irregular octahedron of the orthorhombic phase rotates under pressure and distorts at 65.5 GPa, 67 GPa, 69.5 GPa, and 70 GPa pressure points, respectively. Moreover, the pressure has a good adjustment effect on the band gap of the cubic and orthorhombic phases. The band gap of the orthorhombic phase changes between the direct band gap and the indirect band gap under pressure. The charge difference density and density of states are calculated to study the type of bonds between internal atoms and the effect of pressure on the distribution of orbital electrons. The study of the Crystal Orbital Hamilton Populations and lattice energy of the electron cloud shows that the pressure has a significant adjustment effect on the strength of the Pb–Br covalent bond and the Cs–Br ionic bond. These results provide a comprehensive reference and guidance for the further research and application of perovskite material pressure engineering.
期刊介绍:
Covering major developments in the field of solid state chemistry and related areas such as ceramics and amorphous materials, the Journal of Solid State Chemistry features studies of chemical, structural, thermodynamic, electronic, magnetic, and optical properties and processes in solids.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
