Omeyma Trimeche, Rania Sakka, Ekram Hajji, Abdelmouhaymen Missaoui, Bilel Ben Amor, Ines Bayar, Sana Abid, Hela Marmouch, Hanen Sayedi, Ines Khochtali
{"title":"Portraying the full picture of Neurofibromatosis-Noonan syndrome: a systematic review of literature.","authors":"Omeyma Trimeche, Rania Sakka, Ekram Hajji, Abdelmouhaymen Missaoui, Bilel Ben Amor, Ines Bayar, Sana Abid, Hela Marmouch, Hanen Sayedi, Ines Khochtali","doi":"10.1136/jmg-2024-110253","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and aims: </strong>Neurofibromatosis-Noonan syndrome (NFNS) is an extremely rare genetic entity combining the clinical phenotype of two conditions: neurofibromatosis type 1 syndrome (NF1) and Noonan syndrome (NS). Nevertheless, many inconsistencies reside in our understanding of this condition, mainly its clinical features and genetic background. Through this systematic review, we aim to shed light on the epidemiological features, the broad clinical spectrum, the underlying genetic defects and the associated comorbidities of NFNS.</p><p><strong>Methods: </strong>Medline, Scopus and Google Scholar were searched for publications on the clinical and genetic features of patients with NFNS. Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines were followed and the study protocol was registered in PROSPERO.</p><p><strong>Results: </strong>Of 951 records screened, 42 were eligible. The mean age at diagnosis was 14.7 years ranging from 0 to 69 years. As for the circumstance of discovery of NFNS, it was dominated by family investigation followed by neurofibromas, facial dysmorphia and short stature (SS). Prematurity was noted in 40.9% of cases. The hallmark features of NFNS at diagnosis were 'café au lait' macules, typical facial dysmorphia of NS, postnatal SS, pectus abnormalities, broad neck and lentigines. Macrocephaly, scoliosis and cardiopathies occurred in 26%, 42.4% and 36.9% of cases, respectively. Tumours were found in 18.4% of cases. As for the genetic foundation of NFNS, <i>NF1</i> gene mutations were depicted in 87.5% of individuals.</p><p><strong>Conclusions: </strong>Based on our findings, we emphasise on the importance of searching for NS features in patients with NF1 since the prognosis, comorbidities and consequently management could be altered.</p><p><strong>Prospero registration number: </strong>42024522238.</p>","PeriodicalId":16237,"journal":{"name":"Journal of Medical Genetics","volume":" ","pages":"109-116"},"PeriodicalIF":3.4000,"publicationDate":"2025-01-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11877060/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1136/jmg-2024-110253","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
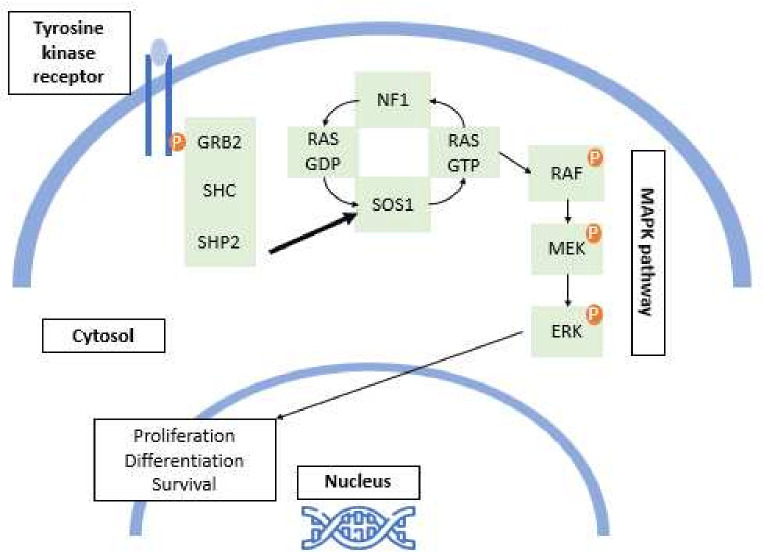
Background and aims: Neurofibromatosis-Noonan syndrome (NFNS) is an extremely rare genetic entity combining the clinical phenotype of two conditions: neurofibromatosis type 1 syndrome (NF1) and Noonan syndrome (NS). Nevertheless, many inconsistencies reside in our understanding of this condition, mainly its clinical features and genetic background. Through this systematic review, we aim to shed light on the epidemiological features, the broad clinical spectrum, the underlying genetic defects and the associated comorbidities of NFNS.
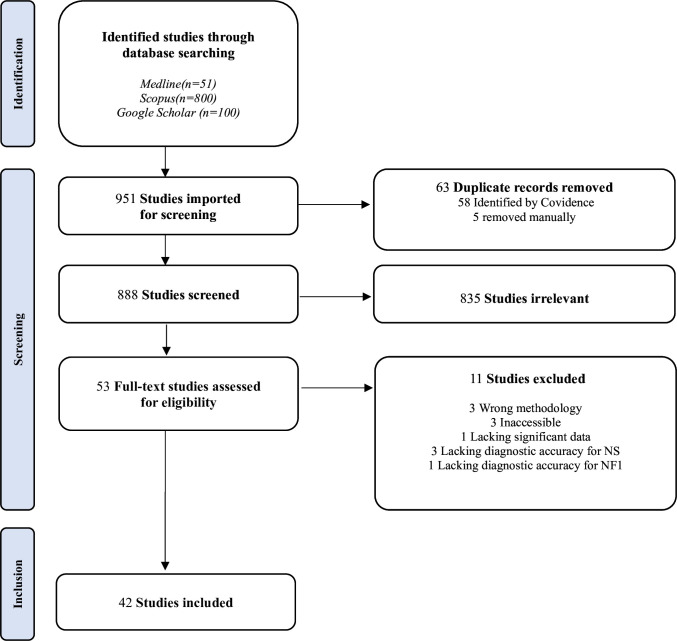
Methods: Medline, Scopus and Google Scholar were searched for publications on the clinical and genetic features of patients with NFNS. Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines were followed and the study protocol was registered in PROSPERO.
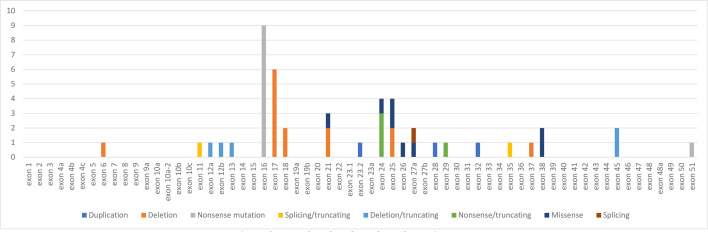
Results: Of 951 records screened, 42 were eligible. The mean age at diagnosis was 14.7 years ranging from 0 to 69 years. As for the circumstance of discovery of NFNS, it was dominated by family investigation followed by neurofibromas, facial dysmorphia and short stature (SS). Prematurity was noted in 40.9% of cases. The hallmark features of NFNS at diagnosis were 'café au lait' macules, typical facial dysmorphia of NS, postnatal SS, pectus abnormalities, broad neck and lentigines. Macrocephaly, scoliosis and cardiopathies occurred in 26%, 42.4% and 36.9% of cases, respectively. Tumours were found in 18.4% of cases. As for the genetic foundation of NFNS, NF1 gene mutations were depicted in 87.5% of individuals.
Conclusions: Based on our findings, we emphasise on the importance of searching for NS features in patients with NF1 since the prognosis, comorbidities and consequently management could be altered.
期刊介绍:
Journal of Medical Genetics is a leading international peer-reviewed journal covering original research in human genetics, including reviews of and opinion on the latest developments. Articles cover the molecular basis of human disease including germline cancer genetics, clinical manifestations of genetic disorders, applications of molecular genetics to medical practice and the systematic evaluation of such applications worldwide.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
